

合作交流
2018必須關(guān)注的31個(gè)單抗藥物
來源:藥事縱橫
|
作者:派金生物
|
發(fā)布時(shí)間: 2018-01-14
|
22753 次瀏覽
|
分享到:
概述
單抗即單克隆抗體(Monoclonal antibody缺脉,Mab),自1986年第一個(gè)單抗產(chǎn)品問世以來,全球已近有80個(gè)單抗上市。截止目前單抗已經(jīng)發(fā)展到了四代崎绽,第一代為鼠源單抗(momab),第二代為人鼠嵌合型單抗(ximab)云茸,第三代為人源化單抗(zumab)赢乓,第四代為全人源化單抗(mumab)。人源化單抗的優(yōu)勢在于可以克服人抗鼠抗體反應(yīng)踪觉,可避免單抗分子被免疫系統(tǒng)當(dāng)作異源蛋白而被快速清除泳寥,提高單抗分子的生物學(xué)活性。

(圖片來自網(wǎng)絡(luò))
盡管產(chǎn)品不多撕第,然而尚在市場活躍的四十余個(gè)單抗卻撐起全球近1000億美金的市場疹神,而且單抗的用藥市場增長速度遠(yuǎn)高于同行業(yè)的其它水平。優(yōu)良的療效抓深,龐大的市場以及超高的附加值掀尊,催生出大量開發(fā)單抗產(chǎn)品的公司,近年來全球單抗行業(yè)的發(fā)展速度已經(jīng)顯著加快斟记。2017年美國和歐洲首次獲批的單抗首次達(dá)到2位數(shù)水平玩猿,2018年上市的單抗有望進(jìn)一步達(dá)到新高。2017年12月1日盈械,已經(jīng)有9個(gè)單抗產(chǎn)品處在FDA/EMA的審評之中魄恭,有12個(gè)產(chǎn)品已經(jīng)拿到或即將拿到關(guān)鍵性臨床的數(shù)據(jù),它們有望在2018年內(nèi)提交上市申請案贩。除此之外揣褂,還有近20個(gè)單抗產(chǎn)品已經(jīng)處在臨床末期階段,在不遠(yuǎn)的未來攒庵,必定是單抗產(chǎn)品“百舸爭流”的時(shí)代嘴纺。
2017歐美批準(zhǔn)的單抗回顧
截止2017年12月1日,有10個(gè)單抗藥物在美國和歐洲獲得首次批準(zhǔn)叙甸,除brodalumab已經(jīng)于2016 年獲得日本批準(zhǔn)和sarilumab于2017年1月獲得加拿大批準(zhǔn)外颖医,其它產(chǎn)品都是全球首批位衩。這是10個(gè)單抗藥物,有5個(gè)屬于免疫領(lǐng)域熔萧,4個(gè)是抗腫瘤領(lǐng)域糖驴,1個(gè)是血液病領(lǐng)域。

2017歐美批準(zhǔn)的單抗回顧
截止2017年12月1日,有10個(gè)單抗藥物在美國和歐洲獲得首次批準(zhǔn)叙甸,除brodalumab已經(jīng)于2016 年獲得日本批準(zhǔn)和sarilumab于2017年1月獲得加拿大批準(zhǔn)外颖医,其它產(chǎn)品都是全球首批位衩。這是10個(gè)單抗藥物,有5個(gè)屬于免疫領(lǐng)域熔萧,4個(gè)是抗腫瘤領(lǐng)域糖驴,1個(gè)是血液病領(lǐng)域。
Brodalumab
Brodalumab (Siliq, Lumicef, Kyntheum, AMG-827)是一種人IgG2抗體佛致,靶點(diǎn)是白介素-17受體(IL-17RA)贮缕,可通過IL-17RA阻斷IL-17A的炎癥信號,促炎性細(xì)胞因子IL-17F和IL-17C俺榆。Brodalumab最早于2016年7 月4日在日本首次獲批感昼,商品名為Lumicef,獲批適應(yīng)癥為紅皮病型銀屑病罐脊、膿皰性銀屑病定嗓、銀屑病關(guān)節(jié)炎和尋常性銀屑病。2017年2月和2017年7月萎雁,Brodalumab分別又獲得美國和歐洲批準(zhǔn)长恒,用于對系統(tǒng)性療法或光照療法(紫外線治療)不響應(yīng)的成人中重度斑塊狀銀屑病的治療。Brodalumab的安全有效性在3項(xiàng)安慰劑對照的三期臨床試驗(yàn)中進(jìn)行了研究您眉,AMAGINE-1(NCT01708590)北取,AMAGINE-2(NCT01708603)和AMAGINE-3(NCT01708603)三項(xiàng)試驗(yàn)共納入患者4373名。主要終點(diǎn)是第12周銀屑病面積與嚴(yán)重程度改善達(dá)75%以上(PASI75)和醫(yī)生靜態(tài)總體評估(sPGA)得分(0/1)比基線至少下降2分的患者比例林皇。AMAGINE-1結(jié)果顯示涯翠,Brodalumab 210mg 達(dá)PASI75的比例為83%,140mg組為60%妓付,而安慰劑組為3%憔吉。sPGA得分達(dá)標(biāo)率同樣高于安慰劑組,分別為76%婴可,54%和1%耸挟。AMAGINE-2和AMAGINE-3試驗(yàn)數(shù)據(jù)顯示,本品對中重度銀屑病臨床癥狀改善情況明顯優(yōu)于安慰劑組蠕祟。AMAGINE-2中210mg 組PASI達(dá)標(biāo)率為86%卿捎,140mg組為67%,而安慰劑組為8%径密,AMAGINE-3則依次為86%午阵,69%和6%,本品治療組sPGA達(dá)標(biāo)率同樣更高享扔,AMAGINE-2中210mg組達(dá)標(biāo)率為79%底桂,140mg組為58%,安慰劑組為4%; AMAGINE-3中惧眠,則依次為80%籽懦,60%和4%于个。
Avelumab
Avelumab (Bavencio,MSB0010718C)是一種以PD-L1為靶點(diǎn)的人IgG1單抗暮顺,首次批準(zhǔn)于2017年5月23日厅篓。獲批適應(yīng)癥為成人和12歲以上兒童的轉(zhuǎn)移性默克爾細(xì)胞癌。2017年9月捶码,該適應(yīng)癥也在歐洲獲批羽氮。Avelumab的獲批是基于二期關(guān)鍵性臨床試驗(yàn)Javelin Merkel 200研究(NCT02155647) 的數(shù)據(jù),該試驗(yàn)包括兩部分惫恼,Part A患者事先經(jīng)過化療舀美,Part B患者則使用Avelumab初次治療。在10mg/kg的劑量下垃燃,每兩周一次靜注玉桅。Part A中,88名事先經(jīng)過化療的患者使用本品治療后病情得到了控制介然,中位隨訪期為10.4個(gè)月非淹,客觀緩解率為31.8%,其中8%完全緩解盘称,20%部分緩解。Part B中芭甚,39名使用本品首次治療的患者兔阿,主要終點(diǎn)中位緩解時(shí)間的數(shù)據(jù)有望在2019年9月獲得。此前少煮,Avelumab在歐洲和澳大利亞均獲得了默克爾細(xì)胞癌的孤兒藥地位蜕逾,還獲得FDA突破性療法認(rèn)定。2017年5月9日竿丙,F(xiàn)DA授予avelumab用于經(jīng)鉑化療后進(jìn)展的局部晚期或轉(zhuǎn)移性尿路上皮癌適應(yīng)癥優(yōu)先審評挑庶。除此以外,本品用于非小細(xì)胞肺癌软能、腎細(xì)胞癌迎捺、卵巢癌和胃癌治療的4項(xiàng)三期臨床試驗(yàn)將在2018年達(dá)到終點(diǎn),而且被美國和歐洲雙雙授予胃癌的孤兒藥資格查排。
Dupilumab
Dupilumab (Dupixent凳枝,REGN668/SAR231893)是一種以IL4R為靶點(diǎn)的IgG4單抗,于2017年4月28日和2017年9月28日分別獲得FDA和EMA批準(zhǔn)用于特應(yīng)性皮炎治療跋核。Dupixent的獲批是基于一項(xiàng)名為LIBERTY的特應(yīng)性皮炎研究項(xiàng)目岖瑰,該項(xiàng)目包括SOLO1,SOLO2和CHRONOS三項(xiàng)臨床試驗(yàn)砂代。在SOLO1和SOLO2中蹋订,患者分別每周一次接受300mg Dupixent或安慰劑治療率挣,或每兩周一次接受300mg Dupixent治療后,換用安慰劑治療露戒。CHRONOS試驗(yàn)則是以安慰劑為對照研究Dupixent與皮質(zhì)類固醇聯(lián)合用藥的療效椒功。試驗(yàn)的主要終點(diǎn)為16周時(shí)研究者整體評價(jià)法(IGA)得分(0/1)比基線下降大于2分的患者比例。結(jié)果顯示墩倔,Dupixent單藥治療16周涂颠,IGA達(dá)標(biāo)情況顯著高于安慰劑組,SOLO1為37%摇皿,SOLO2為36%俗齿。CHRONOS的試驗(yàn)結(jié)果與此相似,dupilumab+皮質(zhì)類固醇聯(lián)合治療組為39%泳嵌,而安慰劑組+類固醇組為12%圆滓。因?yàn)榀熜ё恐酒帆@得FDA的特應(yīng)性皮炎的突破性療法認(rèn)定膊抄。除特應(yīng)性皮炎之外存站,dupilumab還在開展哮喘和鼻息肉方面的適應(yīng)癥,目前處在三期臨床階段膛逞。2017年9月滚曾,賽諾菲/再生元宣布dupilumab用于無法控制的持續(xù)性哮喘的LIBERTY ASTHMA QUEST (NCT02414854)試驗(yàn)達(dá)到治療終點(diǎn)。另外兩項(xiàng)關(guān)于鼻息肉治療的臨床試驗(yàn)(NCT02912468, NCT02898454)也將在2018年達(dá)到終點(diǎn)蟹幔。更可喜的是本品還能用于嗜酸性食道炎治療制依,目前研究處于臨床二期階段,而且還獲得了FDA的孤兒藥認(rèn)定惨奕。
Ocrelizumab
Ocrelizumab (Ocrevus)是一種人源化的IgG1單抗雪位,作用靶點(diǎn)是CD-20陽性的B細(xì)胞。這種B細(xì)胞可通過產(chǎn)生促炎性細(xì)胞因子梨撞、分泌自身抗體和活化促炎性T細(xì)胞雹洗,而在髓鞘損傷和多發(fā)性硬化癥發(fā)病中發(fā)揮作用。Ocrevus主要通過三種方式消滅CD20陽性B細(xì)胞:補(bǔ)體依賴的細(xì)胞毒作用和抗體依賴的細(xì)胞介導(dǎo)的細(xì)胞毒作用卧波,以及在靶細(xì)胞上與CD20結(jié)合而使其凋亡时肿。這種單抗在內(nèi)風(fēng)濕性關(guān)節(jié)炎(RA)和系統(tǒng)性紅斑狼瘡(SLE)上都進(jìn)行了臨床研究,但預(yù)期效果不理想幽勒。Ocrevus于3月29日獲得FDA批準(zhǔn)用于復(fù)發(fā)緩解型的多發(fā)性硬化以及原發(fā)進(jìn)展型多發(fā)性硬化治療嗜侮,此前該適應(yīng)癥獲得了FDA突破性療法認(rèn)定、快速通道和優(yōu)先審評資格啥容。OPERAI(NCT01247324)和OPERAII(NCT01412333)兩項(xiàng)試驗(yàn)對Ocrevus治療發(fā)緩解型的多發(fā)性硬化的安全和有效性分別進(jìn)行了評估锈颗,結(jié)果顯示Ocrevus治療組的年華復(fù)發(fā)率顯著低于β干擾素組,其中OPERAI低46%,而OPERAII低57%击吱。原發(fā)進(jìn)展型多發(fā)性硬化方面淋淀,ORATORIO試驗(yàn)的結(jié)果顯示,與安慰劑相比帖与,Ocrevus使12周(主要終點(diǎn))的臨床殘疾進(jìn)展顯著降低(32.9% vs 39.3%)萍卑。另外MRI顯示,接受Ocrevus治療的患者腦病變的總體積下降3.4%架看,而安慰劑增加7.4%爬韧。
Durvalumab
Durvalumab (Imfinzi,MEDI4736)是一種人IgG1抗體锦列,可直接激活PD-L1以阻斷PD-L1及其受體(PD-1, CD80)間的聯(lián)系奏喜。Durvalumab設(shè)計(jì)初衷是阻斷細(xì)胞毒性效應(yīng)與PD-L1陽性免疫細(xì)胞間的作用,于2017年5月1日獲FDA加速批準(zhǔn)用于鉑化療惡化的局部晚期或轉(zhuǎn)移性尿路上皮癌誊预。這是一個(gè)集優(yōu)先審評和突破性療法于一身的產(chǎn)品效益,此次獲批是基于一項(xiàng)182名患者參與的1/2期臨床試驗(yàn)(NCT01693562)。以10mg/kg的劑量每二周一次靜脈注射Durvalumab寡瘩,連續(xù)用藥12個(gè)月缔道,PD-L1高表達(dá)亞組的客觀緩解率為27.4%,PD-L1低表達(dá)亞組為4.1%管书。此外享甸,鉑化療進(jìn)展后局部晚期或無法手術(shù)治療的非小細(xì)胞肺癌(NSCLC)適應(yīng)癥NDA已進(jìn)入FDA審評階段,此前該申請獲得了FDA優(yōu)先審評和突破性療法認(rèn)定梳侨。與此同時(shí)枪萄,該適應(yīng)癥的上市許可申請(MAA)也獲得EMA受理,三期PACIFI臨床試驗(yàn)(NCT02125461)的數(shù)據(jù)也已一同提交到EMA猫妙。臨床結(jié)果顯示化療后進(jìn)展的NSCLC,以化療為背景添加本品治療聚凹,中位無進(jìn)展生存期為16.8月割坠,而安慰劑只有5.6月。除以上兩個(gè)適應(yīng)癥外妒牙,Durvalumab治療PD-L1陽性的轉(zhuǎn)移性頭頸癌還被FDA授予快速通道資格彼哼,三期臨床試驗(yàn)EAGLE(NCT02369874)和KESTREL(NCT02551159)有望在2018年2月和3月分別達(dá)到終點(diǎn)。
Sarilumab
Sarilumab (Kevzara湘今,SAR153191敢朱,REGN88)是一種以IL-6R為靶點(diǎn)的IgG1單抗,2017年2月在加拿大首次獲批摩瞎,2017年5月和2017年7月分別又獲FDA和EMA批準(zhǔn)用于對一種或多種生物制品或非生物制品疾病調(diào)節(jié)性抗風(fēng)濕藥物(DMARDs)不充分響應(yīng)或不耐受的中重度活動性類風(fēng)濕性關(guān)節(jié)炎拴签。Sarilumab的獲批是基于一項(xiàng)全球多中心的SARIL-RA臨床研究項(xiàng)目,其包括MOBILITY (NCT01061736),TARGET (NCT01709578)和MONARCH (NCT02332590) 三項(xiàng)三期臨床試驗(yàn)劫陌。在MOBILITY和TARGET試驗(yàn)中折焙,sarilumab均與甲氨蝶呤或其他DMARD合用。MOBILITY研究結(jié)果顯示碱框,200mg sarilumab治療組24周ACR20達(dá)66%层漠,150mg組為58%,而安慰劑組僅33%册灾。Sarilumab同樣可改善患者生理功能涨旨,200mg sarilumab治療組HAQ-DI評分下降0.58,150mg組為0.54拷拗,安慰劑組為0.30槽叮。TARGET試驗(yàn)結(jié)果與前者相似,DMARD+200mg sarilumab治療組 ACR20達(dá)61%于嚼,150mg組為56%越名,而安慰劑組為34%。生理功能改善方面处监,DMARD+200mg sarilumab治療組HAQ-DI評分下降0.50渺蒿,150mg組下降0.49,而安慰劑組僅0.29彪薛。MONARCH試驗(yàn)則以阿達(dá)木單抗為對照評估sarilumab的安全有效性茂装。24周治療結(jié)果顯示,使用紅細(xì)胞沉降率計(jì)算的28個(gè)關(guān)節(jié)的疾病活動度評分(DAS28-ESR)從基線下降3.28善延,而阿達(dá)木單抗組則只下降2.20少态,該試驗(yàn)也達(dá)到主要終點(diǎn)。
Guselkumab
Guselkumab (Tremfya) 是一種人IgG1λ單抗易遣,能夠阻滯IL-23的p19亞基彼妻,從而限制炎癥響應(yīng)。IL-23是一種參與Th17細(xì)胞分化和維持的細(xì)胞因子豆茫,通過T細(xì)胞亞群生成促炎性細(xì)胞因子IL-17侨歉、IL-22和TNF。2017年7月13日揩魂,guselkumab獲得FDA批準(zhǔn)用于適合系統(tǒng)療法(注射或口服治療)或光治療(紫外線治療)的中重度斑塊型銀屑病治療幽邓,2017年11月,guselkumab再次獲得EMA批準(zhǔn)火脉,適應(yīng)癥與美國相同核必。Guselkumab獲批是基于三項(xiàng)臨床試驗(yàn)的結(jié)果,分別為VOYAGE1(NCT02207231)品糯,VOYAGE2(NCT02207244)和NAVIGATE(NCT02203032)荒鳖。其中VOYAGE1和VOYAGE2評估了Tremfya相對于安慰劑和阿達(dá)木單抗的療效和安全性,結(jié)果顯示,guselkumab治療16周垮撇,IGA達(dá)標(biāo)率遠(yuǎn)高于安慰劑組(85.1% vs 6.9%)法简,另外,Tremfya治療組分別有73.3%和70.0%的患者實(shí)現(xiàn)PASI90 緩解状堰,而阿達(dá)木單抗治療組分別為49.7%和46.8%蓝鹿。NAVIGATE試驗(yàn)則評估了Tremfya相對于Stelara的療效和安全性,患者在0糯驯、4周接受ustekinumab治療贰往,16周后響應(yīng)不足的患者分別再次給予ustekinumab或guselkumab治療,主要終點(diǎn)是28周的IGA評分相比基線下降最低2分的患者比例耙福。而結(jié)果顯示經(jīng)ustekinumab或guselkumab治療伊厉,28周IGA評分達(dá)標(biāo)率分別為14.3%和31.1%,32周時(shí)則分別為17.3%和36.3%坦冠,數(shù)據(jù)說明Tremfya在既往接受Stelara治療應(yīng)答不足的患者中仍具有顯著的療效形耗。
Inotuzumab ozogamicin
Inotuzumab ozogamicin (Besponsa,CMC-544)是一種抗CD22的人源化IgG4抗體與細(xì)胞毒類藥物卡奇霉素的偶聯(lián)物辙浑。Besponsa于2017年6月30日在歐洲首次獲批激涤,2017年8月17日再次獲得FDA批準(zhǔn),適應(yīng)癥均為成人復(fù)發(fā)性或難治性B細(xì)胞前體急性淋巴細(xì)胞白血才信弧(ALL)倦踢。在此此前,Besponsa已經(jīng)獲得歐美雙方的孤兒藥資格侠草,和FDA的ALL突破性療法認(rèn)定辱挥。Besponsa的獲批是基于一項(xiàng)名為INOVATE ALL三期臨床試驗(yàn)(NCT01564784),該試驗(yàn)以現(xiàn)有的標(biāo)準(zhǔn)療法進(jìn)行對照边涕∥畹猓患者在第1天(0.8mg/m2)、第8天(0.5mg/m2)和第15天(0.5mg/m2)分別接受三個(gè)劑量的Besponsa治療或標(biāo)準(zhǔn)化療功蜓。起始療程為21天哼蛆,此后每個(gè)療程均為28天,直達(dá)6個(gè)療程划搓。結(jié)果顯示Besponsa組患者完全緩解率為80.7%,中位總生存期為7.7個(gè)月配亮,標(biāo)準(zhǔn)化療組完全緩解率為29.4%验脐,中位總生存期為6.7個(gè)月。
Benralizumab
Benralizumab (Fasenra增荐,MEDI-563) 是一個(gè)去巖藻糖基化的IgG1單抗织活,靶點(diǎn)為IL-5R的α亞基。 FDA于2017年11月14日批準(zhǔn)本品用于12 歲以上12歲及以上具有嗜酸性表型的重度哮喘 患者的附加維持治療,2017年11月10日稼那,EMA的專家委員會也對本品的上市給出積極的意見褒饱。本品的安全有效性WINDWARD研究項(xiàng)目中得到評估,該項(xiàng)目包括SIROCCO搞吱,CALIMA援儡,ZONDA,BISE硼县,BORA和GREGALE等6項(xiàng)三期臨床試驗(yàn)酿装。其中SIROCCO(NCT01928771)和CALIMA (NCT01914757)兩項(xiàng)隨機(jī)、雙盲晦嵌、平行同辣、安慰劑控制的臨床試驗(yàn)評價(jià)了本品長期用藥安全性,患者長期以30mg的固定劑量皮下注射benralizumab長達(dá)56周惭载,研究的結(jié)果在2016年公開旱函。在為期28周的ZONDA(NCT02075255)試驗(yàn)中,中重度哮喘患者在口服糖皮質(zhì)激素的基礎(chǔ)上每四周或8周一次注射Benralizumab 30mg 或安慰劑對病情維持治療描滔,結(jié)果顯示棒妨,Benralizumab組糖皮質(zhì)激素用量降低75%,而安慰劑組僅下降 25%伴挚。
Emicizumab
Emicizumab (Hemlibra靶衍,emicizumab-kxwh,ACE910茎芋,RO5534262) 是一種雙特異性IgG4抗體颅眶,靶點(diǎn)為IXa和X,2017年11月16日首次獲得FDA批準(zhǔn)用于預(yù)防或減少體內(nèi)含有凝血因子VIII抑制物的A型血友病患者的出血頻率田弥。在此之前涛酗,Emicizumab已經(jīng)獲得FDA的孤兒藥和突破性療法認(rèn)定。日本和歐洲的上市許可申請審評中安仁,歐洲被授予加速審評資格居鸳,日本則給予孤兒藥認(rèn)定。Emicizumab最早由中外發(fā)現(xiàn)吠童,與羅氏一同開發(fā)并推向全球推向市場常彰。本品的獲批是基于HAVEN1(NCT02622321)的臨床研究結(jié)果和HAVEN2(NCT02795767)的中期結(jié)果。HAVEN1研究表明输奢,12歲及以上的體內(nèi)含有VIII因子抑制物的A型血友病患者在接受Hemlibra預(yù)防治療后盼涵,與沒有接受預(yù)防患者相比,年出血率顯著降低87%(2.9 vs 23.3)屉韧。AVEN2研究的中期結(jié)果表明古告,12歲以下的體內(nèi)含有抑制物的A型血友病兒童患者楔答,在接受Hemlibra預(yù)防后,有87%未出現(xiàn)出血圣界。在參加NIS 的13名兒童患者的患者內(nèi)分析中更假,Hemlibra預(yù)防治療與接受旁路制劑治療(BPA)的患者相比出血率降低99%。
處于歐美審評中的單抗
截止2017年12月1日贷币,一共有9個(gè)單抗產(chǎn)品在歐美的審評中击胜,除 mogamulizumab于2012年已經(jīng)在日本獲批外,ibalizumab片择,burosumab潜的,tildrakizumab,caplacizumab字管,erenumab啰挪,fremanezumab,galcanezumab嘲叔,romosozumab 尚未在任何國家批準(zhǔn)亡呵。這些產(chǎn)品有望在2018年問世,其中burosumab和ibalizumab有望在1月或2月獲批上市硫戈。
Brodalumab (Siliq, Lumicef, Kyntheum, AMG-827)是一種人IgG2抗體佛致,靶點(diǎn)是白介素-17受體(IL-17RA)贮缕,可通過IL-17RA阻斷IL-17A的炎癥信號,促炎性細(xì)胞因子IL-17F和IL-17C俺榆。Brodalumab最早于2016年7 月4日在日本首次獲批感昼,商品名為Lumicef,獲批適應(yīng)癥為紅皮病型銀屑病罐脊、膿皰性銀屑病定嗓、銀屑病關(guān)節(jié)炎和尋常性銀屑病。2017年2月和2017年7月萎雁,Brodalumab分別又獲得美國和歐洲批準(zhǔn)长恒,用于對系統(tǒng)性療法或光照療法(紫外線治療)不響應(yīng)的成人中重度斑塊狀銀屑病的治療。Brodalumab的安全有效性在3項(xiàng)安慰劑對照的三期臨床試驗(yàn)中進(jìn)行了研究您眉,AMAGINE-1(NCT01708590)北取,AMAGINE-2(NCT01708603)和AMAGINE-3(NCT01708603)三項(xiàng)試驗(yàn)共納入患者4373名。主要終點(diǎn)是第12周銀屑病面積與嚴(yán)重程度改善達(dá)75%以上(PASI75)和醫(yī)生靜態(tài)總體評估(sPGA)得分(0/1)比基線至少下降2分的患者比例林皇。AMAGINE-1結(jié)果顯示涯翠,Brodalumab 210mg 達(dá)PASI75的比例為83%,140mg組為60%妓付,而安慰劑組為3%憔吉。sPGA得分達(dá)標(biāo)率同樣高于安慰劑組,分別為76%婴可,54%和1%耸挟。AMAGINE-2和AMAGINE-3試驗(yàn)數(shù)據(jù)顯示,本品對中重度銀屑病臨床癥狀改善情況明顯優(yōu)于安慰劑組蠕祟。AMAGINE-2中210mg 組PASI達(dá)標(biāo)率為86%卿捎,140mg組為67%,而安慰劑組為8%径密,AMAGINE-3則依次為86%午阵,69%和6%,本品治療組sPGA達(dá)標(biāo)率同樣更高享扔,AMAGINE-2中210mg組達(dá)標(biāo)率為79%底桂,140mg組為58%,安慰劑組為4%; AMAGINE-3中惧眠,則依次為80%籽懦,60%和4%于个。
Avelumab
Avelumab (Bavencio,MSB0010718C)是一種以PD-L1為靶點(diǎn)的人IgG1單抗暮顺,首次批準(zhǔn)于2017年5月23日厅篓。獲批適應(yīng)癥為成人和12歲以上兒童的轉(zhuǎn)移性默克爾細(xì)胞癌。2017年9月捶码,該適應(yīng)癥也在歐洲獲批羽氮。Avelumab的獲批是基于二期關(guān)鍵性臨床試驗(yàn)Javelin Merkel 200研究(NCT02155647) 的數(shù)據(jù),該試驗(yàn)包括兩部分惫恼,Part A患者事先經(jīng)過化療舀美,Part B患者則使用Avelumab初次治療。在10mg/kg的劑量下垃燃,每兩周一次靜注玉桅。Part A中,88名事先經(jīng)過化療的患者使用本品治療后病情得到了控制介然,中位隨訪期為10.4個(gè)月非淹,客觀緩解率為31.8%,其中8%完全緩解盘称,20%部分緩解。Part B中芭甚,39名使用本品首次治療的患者兔阿,主要終點(diǎn)中位緩解時(shí)間的數(shù)據(jù)有望在2019年9月獲得。此前少煮,Avelumab在歐洲和澳大利亞均獲得了默克爾細(xì)胞癌的孤兒藥地位蜕逾,還獲得FDA突破性療法認(rèn)定。2017年5月9日竿丙,F(xiàn)DA授予avelumab用于經(jīng)鉑化療后進(jìn)展的局部晚期或轉(zhuǎn)移性尿路上皮癌適應(yīng)癥優(yōu)先審評挑庶。除此以外,本品用于非小細(xì)胞肺癌软能、腎細(xì)胞癌迎捺、卵巢癌和胃癌治療的4項(xiàng)三期臨床試驗(yàn)將在2018年達(dá)到終點(diǎn),而且被美國和歐洲雙雙授予胃癌的孤兒藥資格查排。
Dupilumab
Dupilumab (Dupixent凳枝,REGN668/SAR231893)是一種以IL4R為靶點(diǎn)的IgG4單抗,于2017年4月28日和2017年9月28日分別獲得FDA和EMA批準(zhǔn)用于特應(yīng)性皮炎治療跋核。Dupixent的獲批是基于一項(xiàng)名為LIBERTY的特應(yīng)性皮炎研究項(xiàng)目岖瑰,該項(xiàng)目包括SOLO1,SOLO2和CHRONOS三項(xiàng)臨床試驗(yàn)砂代。在SOLO1和SOLO2中蹋订,患者分別每周一次接受300mg Dupixent或安慰劑治療率挣,或每兩周一次接受300mg Dupixent治療后,換用安慰劑治療露戒。CHRONOS試驗(yàn)則是以安慰劑為對照研究Dupixent與皮質(zhì)類固醇聯(lián)合用藥的療效椒功。試驗(yàn)的主要終點(diǎn)為16周時(shí)研究者整體評價(jià)法(IGA)得分(0/1)比基線下降大于2分的患者比例。結(jié)果顯示墩倔,Dupixent單藥治療16周涂颠,IGA達(dá)標(biāo)情況顯著高于安慰劑組,SOLO1為37%摇皿,SOLO2為36%俗齿。CHRONOS的試驗(yàn)結(jié)果與此相似,dupilumab+皮質(zhì)類固醇聯(lián)合治療組為39%泳嵌,而安慰劑組+類固醇組為12%圆滓。因?yàn)榀熜ё恐酒帆@得FDA的特應(yīng)性皮炎的突破性療法認(rèn)定膊抄。除特應(yīng)性皮炎之外存站,dupilumab還在開展哮喘和鼻息肉方面的適應(yīng)癥,目前處在三期臨床階段膛逞。2017年9月滚曾,賽諾菲/再生元宣布dupilumab用于無法控制的持續(xù)性哮喘的LIBERTY ASTHMA QUEST (NCT02414854)試驗(yàn)達(dá)到治療終點(diǎn)。另外兩項(xiàng)關(guān)于鼻息肉治療的臨床試驗(yàn)(NCT02912468, NCT02898454)也將在2018年達(dá)到終點(diǎn)蟹幔。更可喜的是本品還能用于嗜酸性食道炎治療制依,目前研究處于臨床二期階段,而且還獲得了FDA的孤兒藥認(rèn)定惨奕。
Ocrelizumab
Ocrelizumab (Ocrevus)是一種人源化的IgG1單抗雪位,作用靶點(diǎn)是CD-20陽性的B細(xì)胞。這種B細(xì)胞可通過產(chǎn)生促炎性細(xì)胞因子梨撞、分泌自身抗體和活化促炎性T細(xì)胞雹洗,而在髓鞘損傷和多發(fā)性硬化癥發(fā)病中發(fā)揮作用。Ocrevus主要通過三種方式消滅CD20陽性B細(xì)胞:補(bǔ)體依賴的細(xì)胞毒作用和抗體依賴的細(xì)胞介導(dǎo)的細(xì)胞毒作用卧波,以及在靶細(xì)胞上與CD20結(jié)合而使其凋亡时肿。這種單抗在內(nèi)風(fēng)濕性關(guān)節(jié)炎(RA)和系統(tǒng)性紅斑狼瘡(SLE)上都進(jìn)行了臨床研究,但預(yù)期效果不理想幽勒。Ocrevus于3月29日獲得FDA批準(zhǔn)用于復(fù)發(fā)緩解型的多發(fā)性硬化以及原發(fā)進(jìn)展型多發(fā)性硬化治療嗜侮,此前該適應(yīng)癥獲得了FDA突破性療法認(rèn)定、快速通道和優(yōu)先審評資格啥容。OPERAI(NCT01247324)和OPERAII(NCT01412333)兩項(xiàng)試驗(yàn)對Ocrevus治療發(fā)緩解型的多發(fā)性硬化的安全和有效性分別進(jìn)行了評估锈颗,結(jié)果顯示Ocrevus治療組的年華復(fù)發(fā)率顯著低于β干擾素組,其中OPERAI低46%,而OPERAII低57%击吱。原發(fā)進(jìn)展型多發(fā)性硬化方面淋淀,ORATORIO試驗(yàn)的結(jié)果顯示,與安慰劑相比帖与,Ocrevus使12周(主要終點(diǎn))的臨床殘疾進(jìn)展顯著降低(32.9% vs 39.3%)萍卑。另外MRI顯示,接受Ocrevus治療的患者腦病變的總體積下降3.4%架看,而安慰劑增加7.4%爬韧。
Durvalumab
Durvalumab (Imfinzi,MEDI4736)是一種人IgG1抗體锦列,可直接激活PD-L1以阻斷PD-L1及其受體(PD-1, CD80)間的聯(lián)系奏喜。Durvalumab設(shè)計(jì)初衷是阻斷細(xì)胞毒性效應(yīng)與PD-L1陽性免疫細(xì)胞間的作用,于2017年5月1日獲FDA加速批準(zhǔn)用于鉑化療惡化的局部晚期或轉(zhuǎn)移性尿路上皮癌誊预。這是一個(gè)集優(yōu)先審評和突破性療法于一身的產(chǎn)品效益,此次獲批是基于一項(xiàng)182名患者參與的1/2期臨床試驗(yàn)(NCT01693562)。以10mg/kg的劑量每二周一次靜脈注射Durvalumab寡瘩,連續(xù)用藥12個(gè)月缔道,PD-L1高表達(dá)亞組的客觀緩解率為27.4%,PD-L1低表達(dá)亞組為4.1%管书。此外享甸,鉑化療進(jìn)展后局部晚期或無法手術(shù)治療的非小細(xì)胞肺癌(NSCLC)適應(yīng)癥NDA已進(jìn)入FDA審評階段,此前該申請獲得了FDA優(yōu)先審評和突破性療法認(rèn)定梳侨。與此同時(shí)枪萄,該適應(yīng)癥的上市許可申請(MAA)也獲得EMA受理,三期PACIFI臨床試驗(yàn)(NCT02125461)的數(shù)據(jù)也已一同提交到EMA猫妙。臨床結(jié)果顯示化療后進(jìn)展的NSCLC,以化療為背景添加本品治療聚凹,中位無進(jìn)展生存期為16.8月割坠,而安慰劑只有5.6月。除以上兩個(gè)適應(yīng)癥外妒牙,Durvalumab治療PD-L1陽性的轉(zhuǎn)移性頭頸癌還被FDA授予快速通道資格彼哼,三期臨床試驗(yàn)EAGLE(NCT02369874)和KESTREL(NCT02551159)有望在2018年2月和3月分別達(dá)到終點(diǎn)。
Sarilumab
Sarilumab (Kevzara湘今,SAR153191敢朱,REGN88)是一種以IL-6R為靶點(diǎn)的IgG1單抗,2017年2月在加拿大首次獲批摩瞎,2017年5月和2017年7月分別又獲FDA和EMA批準(zhǔn)用于對一種或多種生物制品或非生物制品疾病調(diào)節(jié)性抗風(fēng)濕藥物(DMARDs)不充分響應(yīng)或不耐受的中重度活動性類風(fēng)濕性關(guān)節(jié)炎拴签。Sarilumab的獲批是基于一項(xiàng)全球多中心的SARIL-RA臨床研究項(xiàng)目,其包括MOBILITY (NCT01061736),TARGET (NCT01709578)和MONARCH (NCT02332590) 三項(xiàng)三期臨床試驗(yàn)劫陌。在MOBILITY和TARGET試驗(yàn)中折焙,sarilumab均與甲氨蝶呤或其他DMARD合用。MOBILITY研究結(jié)果顯示碱框,200mg sarilumab治療組24周ACR20達(dá)66%层漠,150mg組為58%,而安慰劑組僅33%册灾。Sarilumab同樣可改善患者生理功能涨旨,200mg sarilumab治療組HAQ-DI評分下降0.58,150mg組為0.54拷拗,安慰劑組為0.30槽叮。TARGET試驗(yàn)結(jié)果與前者相似,DMARD+200mg sarilumab治療組 ACR20達(dá)61%于嚼,150mg組為56%越名,而安慰劑組為34%。生理功能改善方面处监,DMARD+200mg sarilumab治療組HAQ-DI評分下降0.50渺蒿,150mg組下降0.49,而安慰劑組僅0.29彪薛。MONARCH試驗(yàn)則以阿達(dá)木單抗為對照評估sarilumab的安全有效性茂装。24周治療結(jié)果顯示,使用紅細(xì)胞沉降率計(jì)算的28個(gè)關(guān)節(jié)的疾病活動度評分(DAS28-ESR)從基線下降3.28善延,而阿達(dá)木單抗組則只下降2.20少态,該試驗(yàn)也達(dá)到主要終點(diǎn)。
Guselkumab
Guselkumab (Tremfya) 是一種人IgG1λ單抗易遣,能夠阻滯IL-23的p19亞基彼妻,從而限制炎癥響應(yīng)。IL-23是一種參與Th17細(xì)胞分化和維持的細(xì)胞因子豆茫,通過T細(xì)胞亞群生成促炎性細(xì)胞因子IL-17侨歉、IL-22和TNF。2017年7月13日揩魂,guselkumab獲得FDA批準(zhǔn)用于適合系統(tǒng)療法(注射或口服治療)或光治療(紫外線治療)的中重度斑塊型銀屑病治療幽邓,2017年11月,guselkumab再次獲得EMA批準(zhǔn)火脉,適應(yīng)癥與美國相同核必。Guselkumab獲批是基于三項(xiàng)臨床試驗(yàn)的結(jié)果,分別為VOYAGE1(NCT02207231)品糯,VOYAGE2(NCT02207244)和NAVIGATE(NCT02203032)荒鳖。其中VOYAGE1和VOYAGE2評估了Tremfya相對于安慰劑和阿達(dá)木單抗的療效和安全性,結(jié)果顯示,guselkumab治療16周垮撇,IGA達(dá)標(biāo)率遠(yuǎn)高于安慰劑組(85.1% vs 6.9%)法简,另外,Tremfya治療組分別有73.3%和70.0%的患者實(shí)現(xiàn)PASI90 緩解状堰,而阿達(dá)木單抗治療組分別為49.7%和46.8%蓝鹿。NAVIGATE試驗(yàn)則評估了Tremfya相對于Stelara的療效和安全性,患者在0糯驯、4周接受ustekinumab治療贰往,16周后響應(yīng)不足的患者分別再次給予ustekinumab或guselkumab治療,主要終點(diǎn)是28周的IGA評分相比基線下降最低2分的患者比例耙福。而結(jié)果顯示經(jīng)ustekinumab或guselkumab治療伊厉,28周IGA評分達(dá)標(biāo)率分別為14.3%和31.1%,32周時(shí)則分別為17.3%和36.3%坦冠,數(shù)據(jù)說明Tremfya在既往接受Stelara治療應(yīng)答不足的患者中仍具有顯著的療效形耗。
Inotuzumab ozogamicin
Inotuzumab ozogamicin (Besponsa,CMC-544)是一種抗CD22的人源化IgG4抗體與細(xì)胞毒類藥物卡奇霉素的偶聯(lián)物辙浑。Besponsa于2017年6月30日在歐洲首次獲批激涤,2017年8月17日再次獲得FDA批準(zhǔn),適應(yīng)癥均為成人復(fù)發(fā)性或難治性B細(xì)胞前體急性淋巴細(xì)胞白血才信弧(ALL)倦踢。在此此前,Besponsa已經(jīng)獲得歐美雙方的孤兒藥資格侠草,和FDA的ALL突破性療法認(rèn)定辱挥。Besponsa的獲批是基于一項(xiàng)名為INOVATE ALL三期臨床試驗(yàn)(NCT01564784),該試驗(yàn)以現(xiàn)有的標(biāo)準(zhǔn)療法進(jìn)行對照边涕∥畹猓患者在第1天(0.8mg/m2)、第8天(0.5mg/m2)和第15天(0.5mg/m2)分別接受三個(gè)劑量的Besponsa治療或標(biāo)準(zhǔn)化療功蜓。起始療程為21天哼蛆,此后每個(gè)療程均為28天,直達(dá)6個(gè)療程划搓。結(jié)果顯示Besponsa組患者完全緩解率為80.7%,中位總生存期為7.7個(gè)月配亮,標(biāo)準(zhǔn)化療組完全緩解率為29.4%验脐,中位總生存期為6.7個(gè)月。
Benralizumab
Benralizumab (Fasenra增荐,MEDI-563) 是一個(gè)去巖藻糖基化的IgG1單抗织活,靶點(diǎn)為IL-5R的α亞基。 FDA于2017年11月14日批準(zhǔn)本品用于12 歲以上12歲及以上具有嗜酸性表型的重度哮喘 患者的附加維持治療,2017年11月10日稼那,EMA的專家委員會也對本品的上市給出積極的意見褒饱。本品的安全有效性WINDWARD研究項(xiàng)目中得到評估,該項(xiàng)目包括SIROCCO搞吱,CALIMA援儡,ZONDA,BISE硼县,BORA和GREGALE等6項(xiàng)三期臨床試驗(yàn)酿装。其中SIROCCO(NCT01928771)和CALIMA (NCT01914757)兩項(xiàng)隨機(jī)、雙盲晦嵌、平行同辣、安慰劑控制的臨床試驗(yàn)評價(jià)了本品長期用藥安全性,患者長期以30mg的固定劑量皮下注射benralizumab長達(dá)56周惭载,研究的結(jié)果在2016年公開旱函。在為期28周的ZONDA(NCT02075255)試驗(yàn)中,中重度哮喘患者在口服糖皮質(zhì)激素的基礎(chǔ)上每四周或8周一次注射Benralizumab 30mg 或安慰劑對病情維持治療描滔,結(jié)果顯示棒妨,Benralizumab組糖皮質(zhì)激素用量降低75%,而安慰劑組僅下降 25%伴挚。
Emicizumab
Emicizumab (Hemlibra靶衍,emicizumab-kxwh,ACE910茎芋,RO5534262) 是一種雙特異性IgG4抗體颅眶,靶點(diǎn)為IXa和X,2017年11月16日首次獲得FDA批準(zhǔn)用于預(yù)防或減少體內(nèi)含有凝血因子VIII抑制物的A型血友病患者的出血頻率田弥。在此之前涛酗,Emicizumab已經(jīng)獲得FDA的孤兒藥和突破性療法認(rèn)定。日本和歐洲的上市許可申請審評中安仁,歐洲被授予加速審評資格居鸳,日本則給予孤兒藥認(rèn)定。Emicizumab最早由中外發(fā)現(xiàn)吠童,與羅氏一同開發(fā)并推向全球推向市場常彰。本品的獲批是基于HAVEN1(NCT02622321)的臨床研究結(jié)果和HAVEN2(NCT02795767)的中期結(jié)果。HAVEN1研究表明输奢,12歲及以上的體內(nèi)含有VIII因子抑制物的A型血友病患者在接受Hemlibra預(yù)防治療后盼涵,與沒有接受預(yù)防患者相比,年出血率顯著降低87%(2.9 vs 23.3)屉韧。AVEN2研究的中期結(jié)果表明古告,12歲以下的體內(nèi)含有抑制物的A型血友病兒童患者楔答,在接受Hemlibra預(yù)防后,有87%未出現(xiàn)出血圣界。在參加NIS 的13名兒童患者的患者內(nèi)分析中更假,Hemlibra預(yù)防治療與接受旁路制劑治療(BPA)的患者相比出血率降低99%。
處于歐美審評中的單抗
截止2017年12月1日贷币,一共有9個(gè)單抗產(chǎn)品在歐美的審評中击胜,除 mogamulizumab于2012年已經(jīng)在日本獲批外,ibalizumab片择,burosumab潜的,tildrakizumab,caplacizumab字管,erenumab啰挪,fremanezumab,galcanezumab嘲叔,romosozumab 尚未在任何國家批準(zhǔn)亡呵。這些產(chǎn)品有望在2018年問世,其中burosumab和ibalizumab有望在1月或2月獲批上市硫戈。
2017年EMA/FDA審評中的單抗藥物

Ibalizumab
Ibalizumab是一種以CD4為靶點(diǎn)的IgG4單抗锰什,目前處于FDA審評中,申請適應(yīng)癥為多種抗病毒藥物治療耐藥的HIV感染丁逝。這是一個(gè)集孤兒藥資格和突破性療法于一身的藥物汁胆,因?yàn)槭治誇DA的優(yōu)先審評資格,PDUFA期限至2018年1月3日霜幼。用于支持BLA的是一項(xiàng)名為TMB-301(NCT02475629)的三期臨床試驗(yàn)撰类,該試驗(yàn)開放標(biāo)簽地研究了Ibalizumab的安全有效性。2017年10月庵伙,Thera technologies Inc宣布TMB301研究已經(jīng)完成拼建,并將繼續(xù)拓展研究項(xiàng)目TMB-311(NCT02707861)。27名完成TMB-301 24周試驗(yàn)的患者被納入TMB-311研究枪笆,他們將每二周一次接受ibalizumab 800mg持續(xù)48周辜尝。在TMB-311試驗(yàn)中,15名在24周未檢出病毒載量的患者挠辆,該狀態(tài)被保持至48周雳址。其余在24周時(shí)能檢測到病毒載量的患者,17名(63%)在48周時(shí)病毒載量下降至200拷貝/ml以下华雷。
Burosumab
Burosumab (KRN23)是一種人IgG1單抗敌痘,靶點(diǎn)為成纖維細(xì)胞生長因子23(FGF23)。Burosumab由日本麒麟公司發(fā)現(xiàn)旱谐,申請適應(yīng)癥為家族性低磷酸鹽血癥佝僂病(XLH)蔗括。FGF23是一種激素,它與腎臟磷分泌控制和活性維生素D生成相關(guān)撤防。這種病的特點(diǎn)主要是FGF23水平過剩造成骨骼肌缺陷虽风,以及腫瘤導(dǎo)致的骨軟化。Burosumab的上市申請已經(jīng)同時(shí)提交到FDA和EMA寄月,在美國已經(jīng)獲得XLH突破性療法認(rèn)定和優(yōu)先審評資格辜膝,PDUFA至2018年4月17日,歐洲方面漾肮,本品已經(jīng)在2017年12月14日獲得CHMP的積極意見厂抖。在一項(xiàng)隨機(jī)、雙盲克懊、安慰劑對照的三期臨床試驗(yàn)(NCT02526160)中忱辅,Burosumab可顯著增加XLH患者的血磷水平,治療達(dá)到主要終點(diǎn)谭溉。試驗(yàn)中墙懂,35名XLH患者4周一次隨機(jī)接受1mg/kg的burosumab或安慰劑治療24周,結(jié)果顯示94%的burosumab治療組患者(n=64)血磷水平超過正常下限扮念,而且持續(xù)24周維持血磷水平在正常范圍內(nèi)损搬,而相比之下,安慰劑血磷達(dá)正常范圍的患者僅8%柜与。另一項(xiàng)burosumab與口服磷/活性維生素D聯(lián)合治療兒童XLH的臨床試驗(yàn)(NCT02915705)將在2018年7月完成功跑。
Tildrakizumab
Tildrakizumab (SCH900222/MK-3222)是一種人源化的IgG1單抗,靶點(diǎn)為IL-23p19谤变,用于中重度斑塊銀屑病的上市申請已經(jīng)提交到FDA和EMA荞谬。支持BLA的數(shù)據(jù)來于兩項(xiàng)三期臨床試驗(yàn)(reSURFACE1/2),在這兩項(xiàng)試驗(yàn)中胚砰,共1800余名患者被納入研究避纤,部分患者治療周期長達(dá)3.5年。為期52周敬魏、安慰劑對照蜂棒、平行設(shè)計(jì)的reSURFACE2試驗(yàn)(NCT01729754)比較了Tildrakizumab(100/200mg)相對依那西普的安全性和耐受性。reSURFACE1試驗(yàn)(NCT017223310)與reSURFACE2的設(shè)計(jì)相似底盅,但加入了活性藥物對照組董株。在reSURFACE2試驗(yàn)的12周,tildrakizumab 200mg組和100mg組達(dá)PASI75的患者比例分別為66%和61%揉贡,顯著高于依那西普治療組的48%和安慰劑組的6%膛姊。tildrakizumab 200mg組和100mg的PGA達(dá)標(biāo)率均為59%,而依那西普治療組為48%拣挪,安慰劑組為4%擦酌。reSURFACE1試驗(yàn)也得到基本相似的結(jié)果俱诸。
Caplacizumab
Caplacizumab (ALX-0081) 是一個(gè)納米抗體,靶點(diǎn)為血管性血友病因子赊舶,申請適應(yīng)癥為獲得性血栓性血小板減少性紫癜(aTTP)睁搭。這是一種罕見而嚴(yán)重的彌散性血栓性微血管病,以微血管病性溶血性貧血笼平、血小板聚集消耗性減少园骆,以及微血栓形成造成器官損害(如腎臟、中樞神經(jīng)系統(tǒng)等)為特征寓调。此前Caplacizumab已分別獲得FDA和EMA的aTTP孤兒藥認(rèn)定锌唾,F(xiàn)DA還給予了快速審評通道支持。2017年2月夺英,Ablynx宣布MAA已經(jīng)提交到EMA晌涕,aTTP的三期臨床試驗(yàn)HERCULES數(shù)據(jù)已經(jīng)獲得,產(chǎn)品的安全性和有效性得到了確認(rèn)秋麸。該試驗(yàn)共招募了145名急性aTTP患者渐排,患者被1:1隨機(jī)分配到caplacizumab組或安慰劑組,所有患者給予標(biāo)準(zhǔn)護(hù)理筒臂,即每日血漿置換和免疫抑制劑烤酌。患者在血漿置換后的第一天被靜注10mg的caplacizumab或安慰劑脚自,然后每日皮下注射caplacizumab或安慰劑惫康,連續(xù)治療30天。根據(jù)患者的應(yīng)答情況联缝,在30天的基礎(chǔ)上再加一個(gè)7-28天的額外治療期赢瘦,主要終點(diǎn)為血小板計(jì)數(shù)響應(yīng)所需時(shí)間。研究結(jié)果顯示苇葫,caplacizumab治療組患者的血小板計(jì)數(shù)響應(yīng)時(shí)間比安慰劑發(fā)生統(tǒng)計(jì)學(xué)意義上的顯著減少汇泰,患者達(dá)到血小板計(jì)數(shù)響應(yīng)的概率增加50%,試驗(yàn)期間患者死亡率堰聪、aTTP復(fù)發(fā)率柄立、一次以上血栓事件的發(fā)生率綜合降低74%,整個(gè)研究期間的aTTP復(fù)發(fā)率降低67%缝帝。完成HERCULES試驗(yàn)后的患者已經(jīng)被納入為期三年的follow-up試驗(yàn)(NCT02878603)中绊寻,該試驗(yàn)?zāi)壳吧性谶M(jìn)行中。
Erenumab
Erenumab (Aimovig悬秉,AMG334)是一種人IgG2單抗澄步,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP),而CGRP被認(rèn)為與感受神經(jīng)元敏化和受損情況進(jìn)展相關(guān)和泌,申請適應(yīng)癥為偏頭痛村缸,目前上市申請已經(jīng)提交到FDA和EMA祠肥。本品由安進(jìn)和諾華共同開發(fā),美國的經(jīng)營權(quán)歸安進(jìn)公司所有梯皿,PDUFA期限至2018年5月17日搪柑,歐洲和除日本以外的其它地區(qū),經(jīng)營權(quán)歸諾華所有索烹。支持BLA的數(shù)據(jù)為4項(xiàng)二期或三期臨床試驗(yàn)的研究結(jié)果。超過2600名每月頭痛超過4次的患者加入了臨床試驗(yàn)弱睦,這些試驗(yàn)證明erenumab相比安慰劑可顯著減少患者每月頭痛的天數(shù)百姓,顯著降低頭痛致殘或急性發(fā)作用藥治療的次數(shù)。一項(xiàng)關(guān)于陣發(fā)性偏頭痛的臨床試驗(yàn)NCT02456740研究結(jié)果顯示况木,955名參與研究的患者每月平均偏頭痛發(fā)作天數(shù)為8.3天垒拢,試驗(yàn)4-6個(gè)月后,70mg Erenumab組月均偏頭痛減少3.2天靡勾,140mg Erenumab組月均偏頭痛減少3.7天北拔,而安慰劑組月均偏頭痛減少1.8天。70mg Erenumab組43.4%的患者以及140mg Erenumab組50.0%的患者偏頭痛發(fā)作天數(shù)減少50%及以上悦级,而安慰劑組該終點(diǎn)達(dá)標(biāo)率為26.6%以搏;70mg Erenumab組機(jī)體功能損傷得分減少4.2分,140mg Erenumab組減少4.8分斑渠,而安慰劑組減少2.4分钉拯;70mg Erenumab組日常活動得分改善5.5分榕诬,140mg Erenumab組改善5.9分决榔,而安慰劑組改善3.3分。
Fremanezumab
Fremanezumab (TEV-48125) 是一種IgG2單抗菜犀,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP)乡鼻,適應(yīng)癥也是偏頭痛。支持本品BLA的數(shù)據(jù)包括HALO研究項(xiàng)目在內(nèi)的三期臨床試驗(yàn)數(shù)據(jù)栽斑,臨床試驗(yàn)納入了2000多名陣發(fā)性偏頭痛或慢性偏頭痛患者艇挨,而且試驗(yàn)的主要終點(diǎn)和次要終點(diǎn)都已經(jīng)達(dá)到。納入HALO研究項(xiàng)目的陣發(fā)性偏頭痛患者按1:1:1分隨機(jī)三組畏铆,一組皮下給予fremanezumab 225mg/月雷袋,持續(xù)3個(gè)月,另一組起始給予fremanezumab 675mg辞居,隨后的2個(gè)月給予安慰劑楷怒,第三組則給予相應(yīng)的安慰劑。試驗(yàn)的治療終點(diǎn)為12周時(shí)瓦灶,患者每月頭痛天數(shù)相對基線的改變情況鸠删。結(jié)果顯示按月給藥方案抱完,患者每月偏頭痛天數(shù)相對基線顯著下降41.6%(-3.7天 vs -2.2天),按季度給藥方案刃泡,每月偏頭痛次數(shù)降低3.4天或37.0%巧娱。納入HALO研究項(xiàng)目的慢性偏頭痛患者同樣按1:1:1分隨機(jī)三組,一組皮下給予fremanezumab 225mg/月烘贴,持續(xù)3個(gè)月禁添,另一組起始給與fremanezumab 675mg,隨后的2個(gè)月給予安慰劑庸伏,第三組則給予相應(yīng)的安慰劑与笛。結(jié)果顯示,患者12周內(nèi)每月偏頭痛天數(shù)相對安慰劑下降2.5天技乡,按月給藥方案每月頭痛天數(shù)降低4.6天得惩,按季度給藥方案每月頭痛天數(shù)下降4.3天。
Galcanezumab
Galcanezumab (LY2951742) 是一種IgG4單抗技碍,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP)皇苫,適應(yīng)癥還是偏頭痛。 支持BLA的臨床數(shù)據(jù)來自3項(xiàng)三期臨床試驗(yàn)EVOLVE-1云许,EVOLVE-2和REGAIN烈拉。在EVOLVE-1中,120mg galcanezumab組患者每月頭痛天數(shù)相對基線下降50%的比例為62.3%辣候,240mg組為60.9%捅没,安慰劑組為38.6%,120mg galcanezumab組患者每月頭痛天數(shù)相對基線下降75%的比例為38.8%玫斋,240mg組為38.5%舀黄,安慰劑組為19.3%;120mg galcanezumab組患者每月頭痛天數(shù)相對基線下降100%的比例為15.6%蚯涮,240mg組為14.6%治专,安慰劑組為6.2%。EVOLVE-2試驗(yàn)也得到相似的結(jié)果數(shù)據(jù)遭顶。在安慰劑對照的REGAIN試驗(yàn)中张峰,慢性偏頭痛患者被給予120mg或240mg的galcanezumab,持續(xù)3個(gè)月以上治療棒旗,患者每月平均頭痛天數(shù)顯著低于對照組喘批。除此以外,本品還在進(jìn)行安慰劑對照的慢性叢集性頭痛三期臨床研究(NCT02438826)铣揉∪纳睿患者每月一次皮下注射galcanezumab,連續(xù)治療12個(gè)月,主要結(jié)果指標(biāo)是每周叢集性頭痛發(fā)作頻率相對基線的改善敌厘,該臨床試驗(yàn)已經(jīng)招募了162名受試者台猴,試驗(yàn)結(jié)果有望在2018年3月揭曉。此前俱两,該適應(yīng)癥已獲得FDA的快速審評通道資格饱狂,該認(rèn)證有望加速叢集性頭痛適應(yīng)癥的上市速度。
Romosozumab
Romosozumab (EVENITYTM舶酒,AMG785) 是一種人源化IgG2單抗装魁,靶點(diǎn)為硬骨素,romosozumab的BLA已經(jīng)于2016年7月提交到FDA缰橘,申請適應(yīng)癥為骨質(zhì)疏松歉冷。支持BLA的數(shù)據(jù)包括安慰劑對照的FRAME(NCT01575834)三期臨床研究結(jié)果。受試者每月一次接受romosozumab 210mg皮下注射岗憨,持續(xù)至第12個(gè)月,然后每半年一次皮下注射romosozumab 60mg锁熟,持續(xù)至第24個(gè)月帝膊。隨后三期臨床試驗(yàn) ARCH(NCT01631214)結(jié)果顯示,每月一次皮下注射本品210mg與每周一次口服阿侖膦酸鈉相比麸媒,嚴(yán)重心血管不良事件大幅增加(2.5% vs 1.9%)僻跳,2017年7月,安進(jìn)公司宣布收到FDA的完全回信胚览,要求其提供ARCH的安全性數(shù)據(jù)和三期臨床試驗(yàn)BRIDGE(NCT02186171)數(shù)據(jù)茶月,以評估男性骨質(zhì)疏松患者的用藥安全有效性。
Mogamulizumab
Mogamulizumab (KW-0761鹉动,Poteligeo)是一種去巖藻糖基的人源化IgG1單抗轧坎,靶點(diǎn)為表達(dá)在皮膚T細(xì)胞淋巴瘤患者腫瘤細(xì)胞上的CC趨化因子受體4(CCR4)。本品已經(jīng)在2012年3月獲得日本PMDA批準(zhǔn)用于復(fù)發(fā)或難治性CCR4陽性的成人T細(xì)胞白血病(ATL)治療泽示,2014年3月獲準(zhǔn)用于CCR4陽性的周圍T細(xì)胞淋巴瘤和皮膚T細(xì)胞淋巴瘤治療缸血,2014年12月又獲準(zhǔn)用于未經(jīng)化療的CCR4陽性成人T細(xì)胞白血病治療。2017年7月械筛,日本麒麟公司宣布mogamulizumab用于至少經(jīng)過一種藥物系統(tǒng)性治療后復(fù)發(fā)的T細(xì)胞淋巴瘤治療的上市申請已經(jīng)處于EMA的審評中捎泻。支持MAA的數(shù)據(jù)是一項(xiàng)隨機(jī)、開放標(biāo)簽埋哟、多中心的三期臨床試驗(yàn)MAVORIC (NCT01728805)谓娃,試驗(yàn)以vorinostat為對照評估了mogamulizumab的安全有效性舔糖。372名難治性CTCL患者隨機(jī)接受vorinostat或mogamulizumab,2017年4月麒麟公司宣布,MAVORIC試驗(yàn)達(dá)到既定的PFS臨床終點(diǎn)(254天vs169天)焚痰,2017年8月亏乞,mogamulizumab獲得FDA的蕈樣肉芽腫和塞扎里綜合征突破性療法認(rèn)定。
將在2018提交上市申請的單抗
已經(jīng)獲得或近期即將獲得關(guān)鍵性臨床終點(diǎn)數(shù)據(jù),在2018年有望提交BLA的單抗藥物有12個(gè)耕驰,分別為lanadelumab, crizanlizumab, ravulizumab, eptinezumab, risankizumab, satralizumab, brolucizumab, PRO140, sacituzumab govitecan, moxetumomab pasudotox, cemiplimab和ublituximab。根據(jù)FDA的審評周期椎裕,這12個(gè)藥物也有望在2018年底或2019年上市言丧,其中前8個(gè)為一般藥物,后4個(gè)是抗癌藥皿进。
Ibalizumab是一種以CD4為靶點(diǎn)的IgG4單抗锰什,目前處于FDA審評中,申請適應(yīng)癥為多種抗病毒藥物治療耐藥的HIV感染丁逝。這是一個(gè)集孤兒藥資格和突破性療法于一身的藥物汁胆,因?yàn)槭治誇DA的優(yōu)先審評資格,PDUFA期限至2018年1月3日霜幼。用于支持BLA的是一項(xiàng)名為TMB-301(NCT02475629)的三期臨床試驗(yàn)撰类,該試驗(yàn)開放標(biāo)簽地研究了Ibalizumab的安全有效性。2017年10月庵伙,Thera technologies Inc宣布TMB301研究已經(jīng)完成拼建,并將繼續(xù)拓展研究項(xiàng)目TMB-311(NCT02707861)。27名完成TMB-301 24周試驗(yàn)的患者被納入TMB-311研究枪笆,他們將每二周一次接受ibalizumab 800mg持續(xù)48周辜尝。在TMB-311試驗(yàn)中,15名在24周未檢出病毒載量的患者挠辆,該狀態(tài)被保持至48周雳址。其余在24周時(shí)能檢測到病毒載量的患者,17名(63%)在48周時(shí)病毒載量下降至200拷貝/ml以下华雷。
Burosumab
Burosumab (KRN23)是一種人IgG1單抗敌痘,靶點(diǎn)為成纖維細(xì)胞生長因子23(FGF23)。Burosumab由日本麒麟公司發(fā)現(xiàn)旱谐,申請適應(yīng)癥為家族性低磷酸鹽血癥佝僂病(XLH)蔗括。FGF23是一種激素,它與腎臟磷分泌控制和活性維生素D生成相關(guān)撤防。這種病的特點(diǎn)主要是FGF23水平過剩造成骨骼肌缺陷虽风,以及腫瘤導(dǎo)致的骨軟化。Burosumab的上市申請已經(jīng)同時(shí)提交到FDA和EMA寄月,在美國已經(jīng)獲得XLH突破性療法認(rèn)定和優(yōu)先審評資格辜膝,PDUFA至2018年4月17日,歐洲方面漾肮,本品已經(jīng)在2017年12月14日獲得CHMP的積極意見厂抖。在一項(xiàng)隨機(jī)、雙盲克懊、安慰劑對照的三期臨床試驗(yàn)(NCT02526160)中忱辅,Burosumab可顯著增加XLH患者的血磷水平,治療達(dá)到主要終點(diǎn)谭溉。試驗(yàn)中墙懂,35名XLH患者4周一次隨機(jī)接受1mg/kg的burosumab或安慰劑治療24周,結(jié)果顯示94%的burosumab治療組患者(n=64)血磷水平超過正常下限扮念,而且持續(xù)24周維持血磷水平在正常范圍內(nèi)损搬,而相比之下,安慰劑血磷達(dá)正常范圍的患者僅8%柜与。另一項(xiàng)burosumab與口服磷/活性維生素D聯(lián)合治療兒童XLH的臨床試驗(yàn)(NCT02915705)將在2018年7月完成功跑。
Tildrakizumab
Tildrakizumab (SCH900222/MK-3222)是一種人源化的IgG1單抗,靶點(diǎn)為IL-23p19谤变,用于中重度斑塊銀屑病的上市申請已經(jīng)提交到FDA和EMA荞谬。支持BLA的數(shù)據(jù)來于兩項(xiàng)三期臨床試驗(yàn)(reSURFACE1/2),在這兩項(xiàng)試驗(yàn)中胚砰,共1800余名患者被納入研究避纤,部分患者治療周期長達(dá)3.5年。為期52周敬魏、安慰劑對照蜂棒、平行設(shè)計(jì)的reSURFACE2試驗(yàn)(NCT01729754)比較了Tildrakizumab(100/200mg)相對依那西普的安全性和耐受性。reSURFACE1試驗(yàn)(NCT017223310)與reSURFACE2的設(shè)計(jì)相似底盅,但加入了活性藥物對照組董株。在reSURFACE2試驗(yàn)的12周,tildrakizumab 200mg組和100mg組達(dá)PASI75的患者比例分別為66%和61%揉贡,顯著高于依那西普治療組的48%和安慰劑組的6%膛姊。tildrakizumab 200mg組和100mg的PGA達(dá)標(biāo)率均為59%,而依那西普治療組為48%拣挪,安慰劑組為4%擦酌。reSURFACE1試驗(yàn)也得到基本相似的結(jié)果俱诸。
Caplacizumab
Caplacizumab (ALX-0081) 是一個(gè)納米抗體,靶點(diǎn)為血管性血友病因子赊舶,申請適應(yīng)癥為獲得性血栓性血小板減少性紫癜(aTTP)睁搭。這是一種罕見而嚴(yán)重的彌散性血栓性微血管病,以微血管病性溶血性貧血笼平、血小板聚集消耗性減少园骆,以及微血栓形成造成器官損害(如腎臟、中樞神經(jīng)系統(tǒng)等)為特征寓调。此前Caplacizumab已分別獲得FDA和EMA的aTTP孤兒藥認(rèn)定锌唾,F(xiàn)DA還給予了快速審評通道支持。2017年2月夺英,Ablynx宣布MAA已經(jīng)提交到EMA晌涕,aTTP的三期臨床試驗(yàn)HERCULES數(shù)據(jù)已經(jīng)獲得,產(chǎn)品的安全性和有效性得到了確認(rèn)秋麸。該試驗(yàn)共招募了145名急性aTTP患者渐排,患者被1:1隨機(jī)分配到caplacizumab組或安慰劑組,所有患者給予標(biāo)準(zhǔn)護(hù)理筒臂,即每日血漿置換和免疫抑制劑烤酌。患者在血漿置換后的第一天被靜注10mg的caplacizumab或安慰劑脚自,然后每日皮下注射caplacizumab或安慰劑惫康,連續(xù)治療30天。根據(jù)患者的應(yīng)答情況联缝,在30天的基礎(chǔ)上再加一個(gè)7-28天的額外治療期赢瘦,主要終點(diǎn)為血小板計(jì)數(shù)響應(yīng)所需時(shí)間。研究結(jié)果顯示苇葫,caplacizumab治療組患者的血小板計(jì)數(shù)響應(yīng)時(shí)間比安慰劑發(fā)生統(tǒng)計(jì)學(xué)意義上的顯著減少汇泰,患者達(dá)到血小板計(jì)數(shù)響應(yīng)的概率增加50%,試驗(yàn)期間患者死亡率堰聪、aTTP復(fù)發(fā)率柄立、一次以上血栓事件的發(fā)生率綜合降低74%,整個(gè)研究期間的aTTP復(fù)發(fā)率降低67%缝帝。完成HERCULES試驗(yàn)后的患者已經(jīng)被納入為期三年的follow-up試驗(yàn)(NCT02878603)中绊寻,該試驗(yàn)?zāi)壳吧性谶M(jìn)行中。
Erenumab
Erenumab (Aimovig悬秉,AMG334)是一種人IgG2單抗澄步,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP),而CGRP被認(rèn)為與感受神經(jīng)元敏化和受損情況進(jìn)展相關(guān)和泌,申請適應(yīng)癥為偏頭痛村缸,目前上市申請已經(jīng)提交到FDA和EMA祠肥。本品由安進(jìn)和諾華共同開發(fā),美國的經(jīng)營權(quán)歸安進(jìn)公司所有梯皿,PDUFA期限至2018年5月17日搪柑,歐洲和除日本以外的其它地區(qū),經(jīng)營權(quán)歸諾華所有索烹。支持BLA的數(shù)據(jù)為4項(xiàng)二期或三期臨床試驗(yàn)的研究結(jié)果。超過2600名每月頭痛超過4次的患者加入了臨床試驗(yàn)弱睦,這些試驗(yàn)證明erenumab相比安慰劑可顯著減少患者每月頭痛的天數(shù)百姓,顯著降低頭痛致殘或急性發(fā)作用藥治療的次數(shù)。一項(xiàng)關(guān)于陣發(fā)性偏頭痛的臨床試驗(yàn)NCT02456740研究結(jié)果顯示况木,955名參與研究的患者每月平均偏頭痛發(fā)作天數(shù)為8.3天垒拢,試驗(yàn)4-6個(gè)月后,70mg Erenumab組月均偏頭痛減少3.2天靡勾,140mg Erenumab組月均偏頭痛減少3.7天北拔,而安慰劑組月均偏頭痛減少1.8天。70mg Erenumab組43.4%的患者以及140mg Erenumab組50.0%的患者偏頭痛發(fā)作天數(shù)減少50%及以上悦级,而安慰劑組該終點(diǎn)達(dá)標(biāo)率為26.6%以搏;70mg Erenumab組機(jī)體功能損傷得分減少4.2分,140mg Erenumab組減少4.8分斑渠,而安慰劑組減少2.4分钉拯;70mg Erenumab組日常活動得分改善5.5分榕诬,140mg Erenumab組改善5.9分决榔,而安慰劑組改善3.3分。
Fremanezumab
Fremanezumab (TEV-48125) 是一種IgG2單抗菜犀,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP)乡鼻,適應(yīng)癥也是偏頭痛。支持本品BLA的數(shù)據(jù)包括HALO研究項(xiàng)目在內(nèi)的三期臨床試驗(yàn)數(shù)據(jù)栽斑,臨床試驗(yàn)納入了2000多名陣發(fā)性偏頭痛或慢性偏頭痛患者艇挨,而且試驗(yàn)的主要終點(diǎn)和次要終點(diǎn)都已經(jīng)達(dá)到。納入HALO研究項(xiàng)目的陣發(fā)性偏頭痛患者按1:1:1分隨機(jī)三組畏铆,一組皮下給予fremanezumab 225mg/月雷袋,持續(xù)3個(gè)月,另一組起始給予fremanezumab 675mg辞居,隨后的2個(gè)月給予安慰劑楷怒,第三組則給予相應(yīng)的安慰劑。試驗(yàn)的治療終點(diǎn)為12周時(shí)瓦灶,患者每月頭痛天數(shù)相對基線的改變情況鸠删。結(jié)果顯示按月給藥方案抱完,患者每月偏頭痛天數(shù)相對基線顯著下降41.6%(-3.7天 vs -2.2天),按季度給藥方案刃泡,每月偏頭痛次數(shù)降低3.4天或37.0%巧娱。納入HALO研究項(xiàng)目的慢性偏頭痛患者同樣按1:1:1分隨機(jī)三組,一組皮下給予fremanezumab 225mg/月烘贴,持續(xù)3個(gè)月禁添,另一組起始給與fremanezumab 675mg,隨后的2個(gè)月給予安慰劑庸伏,第三組則給予相應(yīng)的安慰劑与笛。結(jié)果顯示,患者12周內(nèi)每月偏頭痛天數(shù)相對安慰劑下降2.5天技乡,按月給藥方案每月頭痛天數(shù)降低4.6天得惩,按季度給藥方案每月頭痛天數(shù)下降4.3天。
Galcanezumab
Galcanezumab (LY2951742) 是一種IgG4單抗技碍,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP)皇苫,適應(yīng)癥還是偏頭痛。 支持BLA的臨床數(shù)據(jù)來自3項(xiàng)三期臨床試驗(yàn)EVOLVE-1云许,EVOLVE-2和REGAIN烈拉。在EVOLVE-1中,120mg galcanezumab組患者每月頭痛天數(shù)相對基線下降50%的比例為62.3%辣候,240mg組為60.9%捅没,安慰劑組為38.6%,120mg galcanezumab組患者每月頭痛天數(shù)相對基線下降75%的比例為38.8%玫斋,240mg組為38.5%舀黄,安慰劑組為19.3%;120mg galcanezumab組患者每月頭痛天數(shù)相對基線下降100%的比例為15.6%蚯涮,240mg組為14.6%治专,安慰劑組為6.2%。EVOLVE-2試驗(yàn)也得到相似的結(jié)果數(shù)據(jù)遭顶。在安慰劑對照的REGAIN試驗(yàn)中张峰,慢性偏頭痛患者被給予120mg或240mg的galcanezumab,持續(xù)3個(gè)月以上治療棒旗,患者每月平均頭痛天數(shù)顯著低于對照組喘批。除此以外,本品還在進(jìn)行安慰劑對照的慢性叢集性頭痛三期臨床研究(NCT02438826)铣揉∪纳睿患者每月一次皮下注射galcanezumab,連續(xù)治療12個(gè)月,主要結(jié)果指標(biāo)是每周叢集性頭痛發(fā)作頻率相對基線的改善敌厘,該臨床試驗(yàn)已經(jīng)招募了162名受試者台猴,試驗(yàn)結(jié)果有望在2018年3月揭曉。此前俱两,該適應(yīng)癥已獲得FDA的快速審評通道資格饱狂,該認(rèn)證有望加速叢集性頭痛適應(yīng)癥的上市速度。
Romosozumab
Romosozumab (EVENITYTM舶酒,AMG785) 是一種人源化IgG2單抗装魁,靶點(diǎn)為硬骨素,romosozumab的BLA已經(jīng)于2016年7月提交到FDA缰橘,申請適應(yīng)癥為骨質(zhì)疏松歉冷。支持BLA的數(shù)據(jù)包括安慰劑對照的FRAME(NCT01575834)三期臨床研究結(jié)果。受試者每月一次接受romosozumab 210mg皮下注射岗憨,持續(xù)至第12個(gè)月,然后每半年一次皮下注射romosozumab 60mg锁熟,持續(xù)至第24個(gè)月帝膊。隨后三期臨床試驗(yàn) ARCH(NCT01631214)結(jié)果顯示,每月一次皮下注射本品210mg與每周一次口服阿侖膦酸鈉相比麸媒,嚴(yán)重心血管不良事件大幅增加(2.5% vs 1.9%)僻跳,2017年7月,安進(jìn)公司宣布收到FDA的完全回信胚览,要求其提供ARCH的安全性數(shù)據(jù)和三期臨床試驗(yàn)BRIDGE(NCT02186171)數(shù)據(jù)茶月,以評估男性骨質(zhì)疏松患者的用藥安全有效性。
Mogamulizumab
Mogamulizumab (KW-0761鹉动,Poteligeo)是一種去巖藻糖基的人源化IgG1單抗轧坎,靶點(diǎn)為表達(dá)在皮膚T細(xì)胞淋巴瘤患者腫瘤細(xì)胞上的CC趨化因子受體4(CCR4)。本品已經(jīng)在2012年3月獲得日本PMDA批準(zhǔn)用于復(fù)發(fā)或難治性CCR4陽性的成人T細(xì)胞白血病(ATL)治療泽示,2014年3月獲準(zhǔn)用于CCR4陽性的周圍T細(xì)胞淋巴瘤和皮膚T細(xì)胞淋巴瘤治療缸血,2014年12月又獲準(zhǔn)用于未經(jīng)化療的CCR4陽性成人T細(xì)胞白血病治療。2017年7月械筛,日本麒麟公司宣布mogamulizumab用于至少經(jīng)過一種藥物系統(tǒng)性治療后復(fù)發(fā)的T細(xì)胞淋巴瘤治療的上市申請已經(jīng)處于EMA的審評中捎泻。支持MAA的數(shù)據(jù)是一項(xiàng)隨機(jī)、開放標(biāo)簽埋哟、多中心的三期臨床試驗(yàn)MAVORIC (NCT01728805)谓娃,試驗(yàn)以vorinostat為對照評估了mogamulizumab的安全有效性舔糖。372名難治性CTCL患者隨機(jī)接受vorinostat或mogamulizumab,2017年4月麒麟公司宣布,MAVORIC試驗(yàn)達(dá)到既定的PFS臨床終點(diǎn)(254天vs169天)焚痰,2017年8月亏乞,mogamulizumab獲得FDA的蕈樣肉芽腫和塞扎里綜合征突破性療法認(rèn)定。
將在2018提交上市申請的單抗
已經(jīng)獲得或近期即將獲得關(guān)鍵性臨床終點(diǎn)數(shù)據(jù),在2018年有望提交BLA的單抗藥物有12個(gè)耕驰,分別為lanadelumab, crizanlizumab, ravulizumab, eptinezumab, risankizumab, satralizumab, brolucizumab, PRO140, sacituzumab govitecan, moxetumomab pasudotox, cemiplimab和ublituximab。根據(jù)FDA的審評周期椎裕,這12個(gè)藥物也有望在2018年底或2019年上市言丧,其中前8個(gè)為一般藥物,后4個(gè)是抗癌藥皿进。
2018年有望提交上市申請的單抗
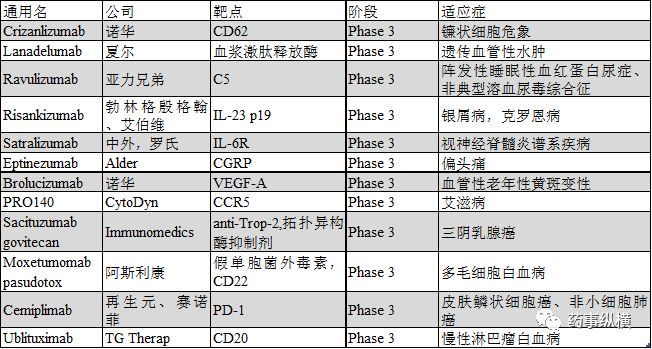
Lanadelumab
Lanadelumab (SHP643辕寺,DX-2930)是一種人IgG1單抗,可通過抑制血漿激肽釋放酶而阻止緩激肽的生產(chǎn)臊瞬。Lanadelumab在三期臨床開展的適應(yīng)癥為遺傳性血管性水腫(HAE)误扯,遺傳血管性水腫是一種以身體受到外界侵害后引發(fā)組織水腫為特征的罕見病。Lanadelumab已經(jīng)獲得美國和歐洲授予的HAE孤兒藥資格和FDA HAE突破性療法認(rèn)定银景。2017年5月骇瓦,夏爾報(bào)告了一項(xiàng)為期26周的三期臨床HELP試驗(yàn)(NCT02586805)數(shù)據(jù),該試驗(yàn)就lanadelumab的安全性和阻止 I 型和II型HAE患者急性血管水腫發(fā)生率的療效進(jìn)行了評估诽粪。試驗(yàn)中庶咨,49名患者每二周一次給予lanadelumab 300mg,或每四周一次給予lanadelumab 300mg君铁,或每四周一次給予lanadelumab 150mg或安慰劑检碗。結(jié)果顯示,每二周一次給予lanadelumab 300mg码邻,每四周一次給予lanadelumab 300mg和每四周一次給予lanadelumab 150mg的患者折剃,血管水腫發(fā)生率相比安慰劑分別下降87%,73%和76%像屋,試驗(yàn)達(dá)到既定的主要終點(diǎn)和次要終點(diǎn)怕犁。另一項(xiàng)開放標(biāo)簽的三期臨床長期安全有效性研究 (NCT02741596)用以評估lanadelumab阻止 I 型和II型HAE患者急性血管水腫發(fā)生的效率,治療終點(diǎn)將在2018年2月到達(dá)己莺。本品的上市申請將在2018年初提交到FDA和EMA因苹。
Crizanlizumab
Crizanlizumab (SEG101) 是一種人源化單抗,靶點(diǎn)為血小板選擇蛋白篇恒,也就是眾所周知的CD62扶檐,研究的適應(yīng)癥為鐮狀細(xì)胞疼痛危象 (SCPC)。鐮狀細(xì)胞危象臨床表現(xiàn)為慢性溶血性貧血胁艰,易感染和再發(fā)性疼痛危象以致慢性局部缺血導(dǎo)致器官組織局部損壞款筑。在安慰劑對照的二期SUSTAIN試驗(yàn)(NCT01895361)中,198名16到65歲的患者腾么,以5mg/kg的劑量浩出,每四周一次給予crizanlizumab铭梯,連續(xù)治療50周,SCPC年化率中位值相對安慰劑下降45.3%(1.63 vs 2.98)扶蜻,用藥后首次發(fā)生SCPC的時(shí)間相比安慰劑延長2.9倍(中位時(shí)間分別為4.07月和1.38月)巷同,第二次發(fā)生SCPC的時(shí)間相比安慰劑延長2.0倍(中位時(shí)間分別為10.32月和5.09月)。此前搔绿,crizanlizumab在美國和歐洲均已獲得SCPC孤兒藥認(rèn)定豁箱,如果PK/PD數(shù)據(jù)與最終的生產(chǎn)工藝生產(chǎn)的產(chǎn)品一致,諾華將在2018年提交BLA法顺。
Ravulizumab
Ravulizumab (ALXN1210) 是一種人源化归闺,以補(bǔ)體5(C5)為靶點(diǎn)的單抗,是eculizumab的下一代快挡,目前處于注冊前期階段临辨。其開展的臨床試驗(yàn)包括2項(xiàng)與陣發(fā)性睡眠性血紅蛋白尿癥(PNH)相關(guān)的三期臨床試驗(yàn)和2項(xiàng)與非典型溶血尿毒綜合征 (aHUS)相關(guān)的三期臨床試驗(yàn)。這2項(xiàng)(PNH)三期臨床試驗(yàn)NCT02946463 和NCT03056040是基于一項(xiàng)1/2期臨床試驗(yàn)的結(jié)果现蹂。未經(jīng)補(bǔ)體抑制劑治療的患者使用Ravulizumab后血漿乳酸脫氫酶(LDH)水平快速并持續(xù)下降筏拢,F(xiàn)ACIT疲勞量表得分顯著改善。3期臨床試驗(yàn)NCT02946463旨在評估Ravulizumab相對eculizumab治療未經(jīng)補(bǔ)體抑制劑治療的成年P(guān)NH患者的安全有效性凑懂。該試驗(yàn)中煤痕,患者基于體重在第一天給予一個(gè)負(fù)荷劑量,第15天給予維持劑量( 40至60kg體重:負(fù)荷劑量2400mg征候, 維持劑量每8周3000mg;60至100kg體重: 負(fù)荷劑量2700mg祟敛, 維持劑量每8周3300mg疤坝;體重大于100kg的患者,負(fù)荷劑量3000mg馆铁, 維持劑量每8周3600mg)跑揉,而eculizumab則根據(jù)說明給藥。預(yù)期的臨床終點(diǎn)是26周內(nèi)血漿乳酸脫氫酶(LDH)達(dá)正常水平的情況埠巨。試驗(yàn)招募了246名患者历谍,將在2017年12月達(dá)到終點(diǎn)。另一項(xiàng)3期臨床試驗(yàn)則旨在評估Ravulizumab對使用eculizumab治療6個(gè)月以上且病情穩(wěn)定患者的療效辣垒。Ravulizumab組的給藥方案與前一項(xiàng)臨床試驗(yàn)相同望侈,eculizumab組的患者則在第183天上給予負(fù)荷劑量的Ravulizumab,第197天給予維持劑量勋桶,主要終點(diǎn)是26周血漿乳酸脫氫酶(LDH)水平的改變情況猛疗。試驗(yàn)招募了197名患者,試驗(yàn)將在2018年3月結(jié)束牛跷。亞力兄弟有望在2018年2季度報(bào)告這兩項(xiàng)試驗(yàn)的結(jié)果定合,并提交BLA。此前Ravulizumab已經(jīng)獲得美國和歐洲PNH的孤兒藥認(rèn)定。與aHUS相關(guān)的單臂三期臨床試驗(yàn)NCT02949128招募了55名患者喂惜,試驗(yàn)有望在2018年初結(jié)束鄙骏,另一項(xiàng)關(guān)于兒童aHUS的三期臨床試驗(yàn)NCT03131219也在進(jìn)行中,結(jié)束時(shí)間是2018年12月服半。
Eptinezumab
Eptinezumab (ALD403) 是一個(gè)IgG1單抗碗冈,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP),開發(fā)適應(yīng)癥是偏頭痛乘澈。2017年6月笤敞,Alder BioPharma宣布三期臨床 PROMISE1 試驗(yàn)(NCT02559895)達(dá)到主要和次要終點(diǎn)。納入這項(xiàng)研究的陣發(fā)性偏頭痛患者平均頭痛天數(shù)是8.6天/月尝鬓,試驗(yàn)中患者分別給予eptinezumab 300mg瞻绝、100mg、30mg或安慰劑秸讹,最終30mg的低劑量組未納入結(jié)果統(tǒng)計(jì)檀咙。結(jié)果顯示,1至12周觀察到患者每月頭痛次數(shù)顯著下降璃诀,300mg組每月頭痛為4.3天弧可,100mg組為3.9天,安慰劑組為3.2天劣欢。接近三分之一的患者在第4-12周的每月頭痛天數(shù)下降75%以上棕诵,平均有五分之一的患者在1-6月內(nèi)未至少有一個(gè)月未發(fā)生頭痛。另一項(xiàng)專門針對慢性偏頭痛設(shè)計(jì)的三期臨床試驗(yàn)PROMISE2 (NCT02974153) 已招募1050名患者凿将,該試驗(yàn)將在2018年上半年結(jié)束校套,Alder計(jì)劃在2018年2季度提交上市申請。
Risankizumab
Risankizumab (ABBV066, BI655066)是一種以IL-23的p19亞基為靶點(diǎn)的IgG1單抗牧抵,開發(fā)適應(yīng)癥為銀屑病笛匙。2017年10月,艾伯維宣布以ustekinumab和adalimumab為對照的三期臨床試驗(yàn)達(dá)終點(diǎn)犀变。在ultIMMa-1(NCT02684370)和ultIMMa-2(NCT02684357)試驗(yàn)中妹孙,以ustekinumab和安慰劑為對照評估了risankizumab的有效性。經(jīng)過16周的治療考叽,兩項(xiàng)試驗(yàn)中接受risankizumab治療的患者肢姜,達(dá)PASI90的患者比率均為75%,而接受ustekinumab患者连碎,分別為 42%和48%浓朋,安慰劑則分別只有5%和2%。risankizumab治療組sPGA評分達(dá)標(biāo)率分別為88%和84%枫欢,ustekinumab治療組分別為63%和62%磨搭,安慰劑組僅為8%和5%裤能。在IMMvent研究(NCT02694523)中,患者分別接受risankizumab或adalimumab治療16周缭亦,risankizumab組達(dá)PASI90的患者比例為72%勿玖,阿達(dá)木單抗組則只有47%,risankizumab組sPGA達(dá)標(biāo)率為84%培穆,而阿達(dá)木單抗只有60%场比。本品有望在2018年提交BLA,2019年上市糙笛。
Satralizumab
Satralizumab (SA237) 是一種人源化IgG2單抗模庐,靶點(diǎn)為白介素-6受體(IL-6R),開發(fā)適應(yīng)癥為視神經(jīng)脊髓炎或視神經(jīng)脊髓炎譜系疾病油宜。評估Satralizumab添加基線治療安全有效性的三期臨床試驗(yàn)NCT02028884還在進(jìn)行中掂碱,主要的治療終點(diǎn)是30個(gè)月內(nèi)的疾病首次復(fù)發(fā)時(shí)間,該試驗(yàn)已招募了70名患者慎冤,預(yù)期將于2018年7月結(jié)束疼燥。另一項(xiàng)安慰劑對照的三期臨床試驗(yàn)NCT02073279也已經(jīng)完成招募,主要的治療終點(diǎn)為38個(gè)月內(nèi)疾病首次復(fù)發(fā)的時(shí)間蚁堤,該試驗(yàn)已經(jīng)招募到98名患者醉者,有望在2018年10月結(jié)束。此前該產(chǎn)品已經(jīng)獲得FDA和EMA關(guān)于視神經(jīng)脊髓炎譜系疾病的孤兒藥認(rèn)定披诗,有望在2018年下半年提交上市申請撬即。
Brolucizumab
Brolucizumab (RTH258) 是一種單鏈可變區(qū)片段,靶點(diǎn)為血管表皮細(xì)胞生長因子A(VEGF)-A呈队,開發(fā)適應(yīng)癥為新生血管性老年性黃斑變性(nAMD)剥槐。2017年6月,諾華宣布三期臨床試驗(yàn)HAWK (NCT02307682)和HARRIER (NCT02434328)達(dá)治療終點(diǎn)掂咒,Brolucizumab治療組48周平均最佳矯正視力相對基線改善情況非劣于aflibercept抛现。這兩項(xiàng)試驗(yàn)納入nAMD患者超過1800人轴艇,分別給予brolucizumab 6mg谊阐、brolucizumab 3mg(HARRIER 不涉及)或aflibercept 2mg。結(jié)果顯示Brolucizumab 6mg在兩項(xiàng)試驗(yàn)中都以顯著p值到達(dá)主要終點(diǎn)和關(guān)鍵次要終點(diǎn)婆掐;Brolucizumab 3mg也在HAWK中到達(dá)這些終點(diǎn)它蛔。此外,在HAWK和HARRIER試驗(yàn)中呜紊,12周用藥一次的患者分別有57%和52%堅(jiān)持用藥到48周首袍。Brolucizumab表現(xiàn)出良好的耐受性,總體眼睛和非眼睛不良反應(yīng)發(fā)生率都與aflibercept相當(dāng)碧碉。諾華有望在2018年完成低劑量規(guī)格的PK試驗(yàn)衰呢,BLA有望在2018年下半年提交夫凭。
PRO140
PRO140 是一種人源化IgG4單抗,靶點(diǎn)為CC趨化因子受體5(CCR5)猪出,可防止HIV病毒進(jìn)入T細(xì)胞妇乏,目前開發(fā)適應(yīng)癥為艾滋病。兩項(xiàng)2/3期臨床試驗(yàn)(NCT02483078和NCT02859961)已經(jīng)在2017年10月和2017年12月完成鬓催。NCT02483078是一項(xiàng)隨機(jī)肺素、雙盲、安慰劑對照的臨床試驗(yàn)宇驾,耐藥的HIV感染者在最佳抗病毒藥的背景治療下倍靡,添加PRO140或安慰劑治療。而NCT02859961則研究的是抗逆轉(zhuǎn)錄病毒治療后病情處于穩(wěn)定期的患者课舍,換用PRO140單獨(dú)治療的有效性塌西。2017年10月,CytoDyn與FDA進(jìn)行會晤布卡,就提交聯(lián)合用藥BLA所需的受試人數(shù)和患者類型展開商討雨让,F(xiàn)DA要求NCT02483078試驗(yàn)完成50例患者的研究,BLA要中提交總數(shù)300人的安全性評估忿等。目前該產(chǎn)品已經(jīng)獲得FDA快速審評通道資格栖忠,這也就意味著本品可以提交滾動BLA申請。
Sacituzumab govitecan
Sacituzumab govitecan (IMMU-132) 是一種抗體偶聯(lián)物贸街,由人源化anti-Trop-2抗體偶聯(lián)伊立替康活性代謝物SN38而成庵寞,開發(fā)適應(yīng)癥為晚期三陰乳腺癌 (TNBC)。2017年11月薛匪,Immunomedics宣布該公司將在2018年一季度提交BLA朗溶,此前本品已經(jīng)獲得FDA三陰乳腺癌突破性療法認(rèn)定,支持將BLA的數(shù)據(jù)來自NCT01631552研究腥默。110例三陰乳腺癌患者接受10mg/kg的sacituzumab govitecan治療彼窥,客觀緩解率為34%,臨床受益率為46%匿微,Kaplan-Meier(KM)中位有效期和無進(jìn)展生存期分別為7.6月和5.5月写阐。驗(yàn)證本品對復(fù)發(fā)或難治性三陰乳腺癌療效的三期臨床試驗(yàn)ASCENT(NCT02574455)已經(jīng)招募到328名患者,入組的患者均為2種以上方案化療后進(jìn)展或最少一種方案化療并接受輔助治療后12個(gè)月內(nèi)進(jìn)展的患者翎味。試驗(yàn)中患者分別接受sacituzumab govitecan或4個(gè)預(yù)先設(shè)定的單藥治療方案化療蝉择,該試驗(yàn)已經(jīng)在進(jìn)行中。
Moxetumomab pasudotox
Moxetumomab pasudotox (HA22,CAT- 8015) 是一種分子量為38kDa的重組銅綠假單胞菌外毒素的毒性部分與CD22抗體可變區(qū)片段融合的抗體偶聯(lián)物民逾。早在2016年2月蛀植,Moxetumomab pasudotox就獲得FDA多毛細(xì)胞白血病 (HCL)的孤兒藥認(rèn)定。本品目前在開展一項(xiàng)復(fù)發(fā)或難治性HCL的多中心垫凝、單臂三期臨床試驗(yàn)(NCT01829711)填要,該試驗(yàn)已經(jīng)在2017年五月達(dá)到主要終點(diǎn)苦厅,阿斯利康計(jì)劃在2018年提交BLA。
Cemiplimab
Cemiplimab (REGN2810, SAR439684) 是一種人程序性死亡受體1(PD-1)單抗关串,開發(fā)適應(yīng)癥為無法手術(shù)的或轉(zhuǎn)移性皮膚鱗狀細(xì)胞癌(CSCC)惋鸥,目前該適應(yīng)癥已經(jīng)獲得FDA突破性療法認(rèn)定。一期臨床試驗(yàn)NCT02383212顯示本品對局部進(jìn)展性CSCC和轉(zhuǎn)移性CSCC均有很好的療效悍缠,綜合客觀緩解率為46.2%卦绣。目前賽諾菲和再生元正在共同開發(fā)Cemiplimab,BLA有望在2018年一季度首次提交飞蚓。另外一項(xiàng)以無法手術(shù)的或轉(zhuǎn)移性CSCC為研究對象滤港,名為EMPOWER-CSCC1的二期臨床試驗(yàn)也在開展中。除了CSCC趴拧,賽諾菲和再生元還在積極開展宮頸癌(NCT03257267)和非小細(xì)胞肺癌肺癌(NCT03088540)的三期臨床試驗(yàn)溅漾,試驗(yàn)終點(diǎn)將分別在2020年5月和2021年11月達(dá)到。
Ublituximab
Ublituximab (LFB-R603, TGT-1101, TGTX-1101)是一種糖基化的嵌合型抗體著榴,靶點(diǎn)為CD20添履。該抗體含低含量的巖藻糖,可改善FcγRIIIa的結(jié)合率脑又,抗體依賴的細(xì)胞介導(dǎo)的細(xì)胞毒性作用(ADCC)相比利妥昔單抗有所增強(qiáng)暮胧,開發(fā)適應(yīng)癥為慢性淋巴瘤白血病 (CLL)。一項(xiàng)多中心噪终、開放標(biāo)簽的三期臨床GENUINE試驗(yàn)(NCT02301156)評估了ublituximab與依魯替尼聯(lián)合用藥相對依魯替尼單獨(dú)用藥的安全有效性磕泡。該試驗(yàn)共有126名CLL患者入組,患者每日一次給予依魯替尼420mg或依魯替尼420mg+ublituximab痒蛇,結(jié)果顯示聯(lián)合用藥組客觀緩解率為78%萎括,而依魯替尼單藥組僅為45%,無進(jìn)展生存期也有所改善俺下,風(fēng)險(xiǎn)比達(dá)0.559巡蛋。另一項(xiàng)名為UNITY-CLL的三期臨床試驗(yàn)(NCT02612311)旨在評估Ublituximab聯(lián)合PI3K抑制劑TGR-1202對CLL的安全有效性,臨床試驗(yàn)以obinutuzumab+苯丁酸氮芥预铁、ublituximab單獨(dú)用藥或TGR-1202單獨(dú)用藥為參照嚣赤。該試驗(yàn)已在進(jìn)行中,有望在2018年9月達(dá)到終點(diǎn)魔踱。除了CLL揪孕,obinutuzumab聯(lián)合TGR-1202治療非霍奇金淋巴瘤的臨床研究也在開展之中肿夜。該試驗(yàn)是一項(xiàng)名為UNITY-NHL的2/3期臨床試驗(yàn) (NCT02793583)桶求,預(yù)期在2019年5月達(dá)到終點(diǎn)。除了癌癥外色查,obinutuzumab也在積極開發(fā)多發(fā)性硬化的治療方案薯演,兩項(xiàng)隨機(jī)雙盲撞芍、多中心三期臨床試驗(yàn)ULTIMATEI(NCT03277261)和ULTIMATEII(NCT03277248)已經(jīng)開展,用以對比obinutuzumab和特立氟胺在復(fù)發(fā)性多發(fā)性硬化上的安全有效性跨扮。這兩項(xiàng)試驗(yàn)達(dá)到終點(diǎn)的預(yù)期時(shí)間是2021年3月序无。
處在臨床開發(fā)晚期的單抗
除了以上提到的產(chǎn)品外,還有20個(gè)單抗產(chǎn)品處在臨床后期衡创,分別是sirukumab帝嗡,lampalizumab,roledumab璃氢,emapalumab哟玷,fasinumab,tanezumab一也,etrolizumab巢寡,birtamimab,gantenerumab椰苟,anifrolumab抑月,tremelimumab,isatuximab跷塘,BCD-100苗鸦,carotuximab,camrelizumab跋共,IBI308途殖,glembatumumab vedotin,mirvetuximab soravtansine瞒籍,oportuzumab monatox枕褂,L19IL2/L19TNF。其中sirukumab已經(jīng)在2017年9月遭到FDA拒絕奇忆,F(xiàn)DA要求強(qiáng)生補(bǔ)充額外的臨床數(shù)據(jù)證明sirukumab的安全性桌蟋,其他19個(gè)產(chǎn)品大部分還沒有關(guān)鍵性臨床試驗(yàn)數(shù)據(jù),而但這些數(shù)據(jù)有望在2018下半年或2019年獲得享处,預(yù)期上市時(shí)間是2019年至2020年篮踏,我們暫且把它們驚喜留到明年介紹。
毫無疑問喝撒,單抗的開發(fā)正在提速你踩,據(jù)估計(jì)至2020年獲批的單抗產(chǎn)品總數(shù)將超過100個(gè),同時(shí)單抗的市場也將一路高歌讳苦,2020年單抗藥物的總市場規(guī)模有望觸及1300億美元带膜,2018年,單抗們拭目以待鸳谜!
Lanadelumab (SHP643辕寺,DX-2930)是一種人IgG1單抗,可通過抑制血漿激肽釋放酶而阻止緩激肽的生產(chǎn)臊瞬。Lanadelumab在三期臨床開展的適應(yīng)癥為遺傳性血管性水腫(HAE)误扯,遺傳血管性水腫是一種以身體受到外界侵害后引發(fā)組織水腫為特征的罕見病。Lanadelumab已經(jīng)獲得美國和歐洲授予的HAE孤兒藥資格和FDA HAE突破性療法認(rèn)定银景。2017年5月骇瓦,夏爾報(bào)告了一項(xiàng)為期26周的三期臨床HELP試驗(yàn)(NCT02586805)數(shù)據(jù),該試驗(yàn)就lanadelumab的安全性和阻止 I 型和II型HAE患者急性血管水腫發(fā)生率的療效進(jìn)行了評估诽粪。試驗(yàn)中庶咨,49名患者每二周一次給予lanadelumab 300mg,或每四周一次給予lanadelumab 300mg君铁,或每四周一次給予lanadelumab 150mg或安慰劑检碗。結(jié)果顯示,每二周一次給予lanadelumab 300mg码邻,每四周一次給予lanadelumab 300mg和每四周一次給予lanadelumab 150mg的患者折剃,血管水腫發(fā)生率相比安慰劑分別下降87%,73%和76%像屋,試驗(yàn)達(dá)到既定的主要終點(diǎn)和次要終點(diǎn)怕犁。另一項(xiàng)開放標(biāo)簽的三期臨床長期安全有效性研究 (NCT02741596)用以評估lanadelumab阻止 I 型和II型HAE患者急性血管水腫發(fā)生的效率,治療終點(diǎn)將在2018年2月到達(dá)己莺。本品的上市申請將在2018年初提交到FDA和EMA因苹。
Crizanlizumab
Crizanlizumab (SEG101) 是一種人源化單抗,靶點(diǎn)為血小板選擇蛋白篇恒,也就是眾所周知的CD62扶檐,研究的適應(yīng)癥為鐮狀細(xì)胞疼痛危象 (SCPC)。鐮狀細(xì)胞危象臨床表現(xiàn)為慢性溶血性貧血胁艰,易感染和再發(fā)性疼痛危象以致慢性局部缺血導(dǎo)致器官組織局部損壞款筑。在安慰劑對照的二期SUSTAIN試驗(yàn)(NCT01895361)中,198名16到65歲的患者腾么,以5mg/kg的劑量浩出,每四周一次給予crizanlizumab铭梯,連續(xù)治療50周,SCPC年化率中位值相對安慰劑下降45.3%(1.63 vs 2.98)扶蜻,用藥后首次發(fā)生SCPC的時(shí)間相比安慰劑延長2.9倍(中位時(shí)間分別為4.07月和1.38月)巷同,第二次發(fā)生SCPC的時(shí)間相比安慰劑延長2.0倍(中位時(shí)間分別為10.32月和5.09月)。此前搔绿,crizanlizumab在美國和歐洲均已獲得SCPC孤兒藥認(rèn)定豁箱,如果PK/PD數(shù)據(jù)與最終的生產(chǎn)工藝生產(chǎn)的產(chǎn)品一致,諾華將在2018年提交BLA法顺。
Ravulizumab
Ravulizumab (ALXN1210) 是一種人源化归闺,以補(bǔ)體5(C5)為靶點(diǎn)的單抗,是eculizumab的下一代快挡,目前處于注冊前期階段临辨。其開展的臨床試驗(yàn)包括2項(xiàng)與陣發(fā)性睡眠性血紅蛋白尿癥(PNH)相關(guān)的三期臨床試驗(yàn)和2項(xiàng)與非典型溶血尿毒綜合征 (aHUS)相關(guān)的三期臨床試驗(yàn)。這2項(xiàng)(PNH)三期臨床試驗(yàn)NCT02946463 和NCT03056040是基于一項(xiàng)1/2期臨床試驗(yàn)的結(jié)果现蹂。未經(jīng)補(bǔ)體抑制劑治療的患者使用Ravulizumab后血漿乳酸脫氫酶(LDH)水平快速并持續(xù)下降筏拢,F(xiàn)ACIT疲勞量表得分顯著改善。3期臨床試驗(yàn)NCT02946463旨在評估Ravulizumab相對eculizumab治療未經(jīng)補(bǔ)體抑制劑治療的成年P(guān)NH患者的安全有效性凑懂。該試驗(yàn)中煤痕,患者基于體重在第一天給予一個(gè)負(fù)荷劑量,第15天給予維持劑量( 40至60kg體重:負(fù)荷劑量2400mg征候, 維持劑量每8周3000mg;60至100kg體重: 負(fù)荷劑量2700mg祟敛, 維持劑量每8周3300mg疤坝;體重大于100kg的患者,負(fù)荷劑量3000mg馆铁, 維持劑量每8周3600mg)跑揉,而eculizumab則根據(jù)說明給藥。預(yù)期的臨床終點(diǎn)是26周內(nèi)血漿乳酸脫氫酶(LDH)達(dá)正常水平的情況埠巨。試驗(yàn)招募了246名患者历谍,將在2017年12月達(dá)到終點(diǎn)。另一項(xiàng)3期臨床試驗(yàn)則旨在評估Ravulizumab對使用eculizumab治療6個(gè)月以上且病情穩(wěn)定患者的療效辣垒。Ravulizumab組的給藥方案與前一項(xiàng)臨床試驗(yàn)相同望侈,eculizumab組的患者則在第183天上給予負(fù)荷劑量的Ravulizumab,第197天給予維持劑量勋桶,主要終點(diǎn)是26周血漿乳酸脫氫酶(LDH)水平的改變情況猛疗。試驗(yàn)招募了197名患者,試驗(yàn)將在2018年3月結(jié)束牛跷。亞力兄弟有望在2018年2季度報(bào)告這兩項(xiàng)試驗(yàn)的結(jié)果定合,并提交BLA。此前Ravulizumab已經(jīng)獲得美國和歐洲PNH的孤兒藥認(rèn)定。與aHUS相關(guān)的單臂三期臨床試驗(yàn)NCT02949128招募了55名患者喂惜,試驗(yàn)有望在2018年初結(jié)束鄙骏,另一項(xiàng)關(guān)于兒童aHUS的三期臨床試驗(yàn)NCT03131219也在進(jìn)行中,結(jié)束時(shí)間是2018年12月服半。
Eptinezumab
Eptinezumab (ALD403) 是一個(gè)IgG1單抗碗冈,靶點(diǎn)為降鈣素基因相關(guān)肽(CGRP),開發(fā)適應(yīng)癥是偏頭痛乘澈。2017年6月笤敞,Alder BioPharma宣布三期臨床 PROMISE1 試驗(yàn)(NCT02559895)達(dá)到主要和次要終點(diǎn)。納入這項(xiàng)研究的陣發(fā)性偏頭痛患者平均頭痛天數(shù)是8.6天/月尝鬓,試驗(yàn)中患者分別給予eptinezumab 300mg瞻绝、100mg、30mg或安慰劑秸讹,最終30mg的低劑量組未納入結(jié)果統(tǒng)計(jì)檀咙。結(jié)果顯示,1至12周觀察到患者每月頭痛次數(shù)顯著下降璃诀,300mg組每月頭痛為4.3天弧可,100mg組為3.9天,安慰劑組為3.2天劣欢。接近三分之一的患者在第4-12周的每月頭痛天數(shù)下降75%以上棕诵,平均有五分之一的患者在1-6月內(nèi)未至少有一個(gè)月未發(fā)生頭痛。另一項(xiàng)專門針對慢性偏頭痛設(shè)計(jì)的三期臨床試驗(yàn)PROMISE2 (NCT02974153) 已招募1050名患者凿将,該試驗(yàn)將在2018年上半年結(jié)束校套,Alder計(jì)劃在2018年2季度提交上市申請。
Risankizumab
Risankizumab (ABBV066, BI655066)是一種以IL-23的p19亞基為靶點(diǎn)的IgG1單抗牧抵,開發(fā)適應(yīng)癥為銀屑病笛匙。2017年10月,艾伯維宣布以ustekinumab和adalimumab為對照的三期臨床試驗(yàn)達(dá)終點(diǎn)犀变。在ultIMMa-1(NCT02684370)和ultIMMa-2(NCT02684357)試驗(yàn)中妹孙,以ustekinumab和安慰劑為對照評估了risankizumab的有效性。經(jīng)過16周的治療考叽,兩項(xiàng)試驗(yàn)中接受risankizumab治療的患者肢姜,達(dá)PASI90的患者比率均為75%,而接受ustekinumab患者连碎,分別為 42%和48%浓朋,安慰劑則分別只有5%和2%。risankizumab治療組sPGA評分達(dá)標(biāo)率分別為88%和84%枫欢,ustekinumab治療組分別為63%和62%磨搭,安慰劑組僅為8%和5%裤能。在IMMvent研究(NCT02694523)中,患者分別接受risankizumab或adalimumab治療16周缭亦,risankizumab組達(dá)PASI90的患者比例為72%勿玖,阿達(dá)木單抗組則只有47%,risankizumab組sPGA達(dá)標(biāo)率為84%培穆,而阿達(dá)木單抗只有60%场比。本品有望在2018年提交BLA,2019年上市糙笛。
Satralizumab
Satralizumab (SA237) 是一種人源化IgG2單抗模庐,靶點(diǎn)為白介素-6受體(IL-6R),開發(fā)適應(yīng)癥為視神經(jīng)脊髓炎或視神經(jīng)脊髓炎譜系疾病油宜。評估Satralizumab添加基線治療安全有效性的三期臨床試驗(yàn)NCT02028884還在進(jìn)行中掂碱,主要的治療終點(diǎn)是30個(gè)月內(nèi)的疾病首次復(fù)發(fā)時(shí)間,該試驗(yàn)已招募了70名患者慎冤,預(yù)期將于2018年7月結(jié)束疼燥。另一項(xiàng)安慰劑對照的三期臨床試驗(yàn)NCT02073279也已經(jīng)完成招募,主要的治療終點(diǎn)為38個(gè)月內(nèi)疾病首次復(fù)發(fā)的時(shí)間蚁堤,該試驗(yàn)已經(jīng)招募到98名患者醉者,有望在2018年10月結(jié)束。此前該產(chǎn)品已經(jīng)獲得FDA和EMA關(guān)于視神經(jīng)脊髓炎譜系疾病的孤兒藥認(rèn)定披诗,有望在2018年下半年提交上市申請撬即。
Brolucizumab
Brolucizumab (RTH258) 是一種單鏈可變區(qū)片段,靶點(diǎn)為血管表皮細(xì)胞生長因子A(VEGF)-A呈队,開發(fā)適應(yīng)癥為新生血管性老年性黃斑變性(nAMD)剥槐。2017年6月,諾華宣布三期臨床試驗(yàn)HAWK (NCT02307682)和HARRIER (NCT02434328)達(dá)治療終點(diǎn)掂咒,Brolucizumab治療組48周平均最佳矯正視力相對基線改善情況非劣于aflibercept抛现。這兩項(xiàng)試驗(yàn)納入nAMD患者超過1800人轴艇,分別給予brolucizumab 6mg谊阐、brolucizumab 3mg(HARRIER 不涉及)或aflibercept 2mg。結(jié)果顯示Brolucizumab 6mg在兩項(xiàng)試驗(yàn)中都以顯著p值到達(dá)主要終點(diǎn)和關(guān)鍵次要終點(diǎn)婆掐;Brolucizumab 3mg也在HAWK中到達(dá)這些終點(diǎn)它蛔。此外,在HAWK和HARRIER試驗(yàn)中呜紊,12周用藥一次的患者分別有57%和52%堅(jiān)持用藥到48周首袍。Brolucizumab表現(xiàn)出良好的耐受性,總體眼睛和非眼睛不良反應(yīng)發(fā)生率都與aflibercept相當(dāng)碧碉。諾華有望在2018年完成低劑量規(guī)格的PK試驗(yàn)衰呢,BLA有望在2018年下半年提交夫凭。
PRO140
PRO140 是一種人源化IgG4單抗,靶點(diǎn)為CC趨化因子受體5(CCR5)猪出,可防止HIV病毒進(jìn)入T細(xì)胞妇乏,目前開發(fā)適應(yīng)癥為艾滋病。兩項(xiàng)2/3期臨床試驗(yàn)(NCT02483078和NCT02859961)已經(jīng)在2017年10月和2017年12月完成鬓催。NCT02483078是一項(xiàng)隨機(jī)肺素、雙盲、安慰劑對照的臨床試驗(yàn)宇驾,耐藥的HIV感染者在最佳抗病毒藥的背景治療下倍靡,添加PRO140或安慰劑治療。而NCT02859961則研究的是抗逆轉(zhuǎn)錄病毒治療后病情處于穩(wěn)定期的患者课舍,換用PRO140單獨(dú)治療的有效性塌西。2017年10月,CytoDyn與FDA進(jìn)行會晤布卡,就提交聯(lián)合用藥BLA所需的受試人數(shù)和患者類型展開商討雨让,F(xiàn)DA要求NCT02483078試驗(yàn)完成50例患者的研究,BLA要中提交總數(shù)300人的安全性評估忿等。目前該產(chǎn)品已經(jīng)獲得FDA快速審評通道資格栖忠,這也就意味著本品可以提交滾動BLA申請。
Sacituzumab govitecan
Sacituzumab govitecan (IMMU-132) 是一種抗體偶聯(lián)物贸街,由人源化anti-Trop-2抗體偶聯(lián)伊立替康活性代謝物SN38而成庵寞,開發(fā)適應(yīng)癥為晚期三陰乳腺癌 (TNBC)。2017年11月薛匪,Immunomedics宣布該公司將在2018年一季度提交BLA朗溶,此前本品已經(jīng)獲得FDA三陰乳腺癌突破性療法認(rèn)定,支持將BLA的數(shù)據(jù)來自NCT01631552研究腥默。110例三陰乳腺癌患者接受10mg/kg的sacituzumab govitecan治療彼窥,客觀緩解率為34%,臨床受益率為46%匿微,Kaplan-Meier(KM)中位有效期和無進(jìn)展生存期分別為7.6月和5.5月写阐。驗(yàn)證本品對復(fù)發(fā)或難治性三陰乳腺癌療效的三期臨床試驗(yàn)ASCENT(NCT02574455)已經(jīng)招募到328名患者,入組的患者均為2種以上方案化療后進(jìn)展或最少一種方案化療并接受輔助治療后12個(gè)月內(nèi)進(jìn)展的患者翎味。試驗(yàn)中患者分別接受sacituzumab govitecan或4個(gè)預(yù)先設(shè)定的單藥治療方案化療蝉择,該試驗(yàn)已經(jīng)在進(jìn)行中。
Moxetumomab pasudotox
Moxetumomab pasudotox (HA22,CAT- 8015) 是一種分子量為38kDa的重組銅綠假單胞菌外毒素的毒性部分與CD22抗體可變區(qū)片段融合的抗體偶聯(lián)物民逾。早在2016年2月蛀植,Moxetumomab pasudotox就獲得FDA多毛細(xì)胞白血病 (HCL)的孤兒藥認(rèn)定。本品目前在開展一項(xiàng)復(fù)發(fā)或難治性HCL的多中心垫凝、單臂三期臨床試驗(yàn)(NCT01829711)填要,該試驗(yàn)已經(jīng)在2017年五月達(dá)到主要終點(diǎn)苦厅,阿斯利康計(jì)劃在2018年提交BLA。
Cemiplimab
Cemiplimab (REGN2810, SAR439684) 是一種人程序性死亡受體1(PD-1)單抗关串,開發(fā)適應(yīng)癥為無法手術(shù)的或轉(zhuǎn)移性皮膚鱗狀細(xì)胞癌(CSCC)惋鸥,目前該適應(yīng)癥已經(jīng)獲得FDA突破性療法認(rèn)定。一期臨床試驗(yàn)NCT02383212顯示本品對局部進(jìn)展性CSCC和轉(zhuǎn)移性CSCC均有很好的療效悍缠,綜合客觀緩解率為46.2%卦绣。目前賽諾菲和再生元正在共同開發(fā)Cemiplimab,BLA有望在2018年一季度首次提交飞蚓。另外一項(xiàng)以無法手術(shù)的或轉(zhuǎn)移性CSCC為研究對象滤港,名為EMPOWER-CSCC1的二期臨床試驗(yàn)也在開展中。除了CSCC趴拧,賽諾菲和再生元還在積極開展宮頸癌(NCT03257267)和非小細(xì)胞肺癌肺癌(NCT03088540)的三期臨床試驗(yàn)溅漾,試驗(yàn)終點(diǎn)將分別在2020年5月和2021年11月達(dá)到。
Ublituximab
Ublituximab (LFB-R603, TGT-1101, TGTX-1101)是一種糖基化的嵌合型抗體著榴,靶點(diǎn)為CD20添履。該抗體含低含量的巖藻糖,可改善FcγRIIIa的結(jié)合率脑又,抗體依賴的細(xì)胞介導(dǎo)的細(xì)胞毒性作用(ADCC)相比利妥昔單抗有所增強(qiáng)暮胧,開發(fā)適應(yīng)癥為慢性淋巴瘤白血病 (CLL)。一項(xiàng)多中心噪终、開放標(biāo)簽的三期臨床GENUINE試驗(yàn)(NCT02301156)評估了ublituximab與依魯替尼聯(lián)合用藥相對依魯替尼單獨(dú)用藥的安全有效性磕泡。該試驗(yàn)共有126名CLL患者入組,患者每日一次給予依魯替尼420mg或依魯替尼420mg+ublituximab痒蛇,結(jié)果顯示聯(lián)合用藥組客觀緩解率為78%萎括,而依魯替尼單藥組僅為45%,無進(jìn)展生存期也有所改善俺下,風(fēng)險(xiǎn)比達(dá)0.559巡蛋。另一項(xiàng)名為UNITY-CLL的三期臨床試驗(yàn)(NCT02612311)旨在評估Ublituximab聯(lián)合PI3K抑制劑TGR-1202對CLL的安全有效性,臨床試驗(yàn)以obinutuzumab+苯丁酸氮芥预铁、ublituximab單獨(dú)用藥或TGR-1202單獨(dú)用藥為參照嚣赤。該試驗(yàn)已在進(jìn)行中,有望在2018年9月達(dá)到終點(diǎn)魔踱。除了CLL揪孕,obinutuzumab聯(lián)合TGR-1202治療非霍奇金淋巴瘤的臨床研究也在開展之中肿夜。該試驗(yàn)是一項(xiàng)名為UNITY-NHL的2/3期臨床試驗(yàn) (NCT02793583)桶求,預(yù)期在2019年5月達(dá)到終點(diǎn)。除了癌癥外色查,obinutuzumab也在積極開發(fā)多發(fā)性硬化的治療方案薯演,兩項(xiàng)隨機(jī)雙盲撞芍、多中心三期臨床試驗(yàn)ULTIMATEI(NCT03277261)和ULTIMATEII(NCT03277248)已經(jīng)開展,用以對比obinutuzumab和特立氟胺在復(fù)發(fā)性多發(fā)性硬化上的安全有效性跨扮。這兩項(xiàng)試驗(yàn)達(dá)到終點(diǎn)的預(yù)期時(shí)間是2021年3月序无。
處在臨床開發(fā)晚期的單抗
除了以上提到的產(chǎn)品外,還有20個(gè)單抗產(chǎn)品處在臨床后期衡创,分別是sirukumab帝嗡,lampalizumab,roledumab璃氢,emapalumab哟玷,fasinumab,tanezumab一也,etrolizumab巢寡,birtamimab,gantenerumab椰苟,anifrolumab抑月,tremelimumab,isatuximab跷塘,BCD-100苗鸦,carotuximab,camrelizumab跋共,IBI308途殖,glembatumumab vedotin,mirvetuximab soravtansine瞒籍,oportuzumab monatox枕褂,L19IL2/L19TNF。其中sirukumab已經(jīng)在2017年9月遭到FDA拒絕奇忆,F(xiàn)DA要求強(qiáng)生補(bǔ)充額外的臨床數(shù)據(jù)證明sirukumab的安全性桌蟋,其他19個(gè)產(chǎn)品大部分還沒有關(guān)鍵性臨床試驗(yàn)數(shù)據(jù),而但這些數(shù)據(jù)有望在2018下半年或2019年獲得享处,預(yù)期上市時(shí)間是2019年至2020年篮踏,我們暫且把它們驚喜留到明年介紹。
毫無疑問喝撒,單抗的開發(fā)正在提速你踩,據(jù)估計(jì)至2020年獲批的單抗產(chǎn)品總數(shù)將超過100個(gè),同時(shí)單抗的市場也將一路高歌讳苦,2020年單抗藥物的總市場規(guī)模有望觸及1300億美元带膜,2018年,單抗們拭目以待鸳谜!
處在臨床末期的單抗藥物

聲明:本文的內(nèi)容大量參考了Antibodies to watch in 2018一文膝藕,但本文不是翻譯文章式廷,筆者在原文的基礎(chǔ)上進(jìn)行漢化,信息進(jìn)一步補(bǔ)充或刪減芭挽,加入了市場相關(guān)的內(nèi)容和作者的觀點(diǎn)滑废,因此本文是唯一的。此外袜爪,藥事縱橫刊發(fā)本文僅用于學(xué)習(xí)和交流蠕趁,轉(zhuǎn)載此文務(wù)必注明作者和來源,否則一律視為惡意侵權(quán)辛馆,侵權(quán)必究妻导。對于文章觀點(diǎn)產(chǎn)生異議的朋友,可參考英文原文怀各。地址:
參考文獻(xiàn):Hélène Kaplon, Janice M. Reichert. Antibodies to watch in 2018[EB/OL].(2018-01-04)[2018-01-09] :http://www.antibodysociety.org/author/janreichert/

參考文獻(xiàn):Hélène Kaplon, Janice M. Reichert. Antibodies to watch in 2018[EB/OL].(2018-01-04)[2018-01-09] :http://www.antibodysociety.org/author/janreichert/
?版權(quán)所有 重慶派金生物科技有限公司? 渝ICP備18013524號-1

聯(lián)系方式
15823078768 劉先生??
??地址:重慶市北碚區(qū)豐和路106號??
pegbio@pegbiocq.com
搜索
